Unraveling the role of regulatory cell death in sepsis: an integrated analysis of bulk and single-cell sequencing data
- Published
- Accepted
- Received
- Academic Editor
- Kazumichi Fujioka
- Subject Areas
- Bioinformatics, Genomics, Immunology, Infectious Diseases, Translational Medicine
- Keywords
- Sepsis, Regulatory cell death, Machine learning, scRNA, CLP
- Copyright
- © 2025 Cao et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2025. Unraveling the role of regulatory cell death in sepsis: an integrated analysis of bulk and single-cell sequencing data. PeerJ 13:e20167 https://doi.org/10.7717/peerj.20167
Abstract
Background
Sepsis is a life-threatening condition characterized by systemic inflammation and dysfunction of multiple organs. Recently, regulatory cell death (RCD) has emerged as a distinct pathological feature and serve as a potential source of biomarkers or therapeutic targets in sepsis.
Methods
Comprehensive transcriptomic datasets of sepsis were accessed from the Gene Expression Omnibus (GEO) database. Genes involved in 18 RCD pathways were compiled from databases and published literature. The limma package was utilized to identify differentially expressed genes (DEGs). The Gene Set Variation Analysis (GSVA), CIBERSORT, Weighted Gene Co-expression Network Analysis (WGCNA), and receiver operating characteristic (ROC) analyses were combined to identify key RCDs pathways. Core RCD-related DEGs (RRDs) were selected using Least Absolute Shrinkage and Selection Operator (LASSO), Support Vector Machine (SVM), and Random Forest (RF) machine learning methods. The expression patterns and diagnostic performance of the core RRDs were validated across multiple datasets and further confirmed through meta-analysis. Immune localization of RRDs was examined using single-cell transcriptomic data. Prognostic significance was evaluated using multivariate Cox analysis. Finally, the mRNA expression level was validated using quantitative real-time polymerase chain reaction (qRT-PCR).
Results
Zinc Finger DHHC-Type Containing 3 (ZDHHC3), Chloride Intracellular Channel 1 (CLIC1), Glutathione S-Transferase Omega 1 (GSTO1), Biogenesis of Lysosomal Organelles Complex 1 Subunit 1 (BLOC1S1), and Toll-Like Receptor 5 (TLR5) were considered as core RRDs, with monocytes and neutrophils serving as the principal cell types responsible for their overexpression and likely contributing critically to their downstream biological effects. Among them, ZDHHC3 and TLR5 were identified as independent risk factors for sepsis. Their significantly elevated mRNA expression in septic mice was confirmed by qRT-PCR.
Conclusion
Findings from this study underscored the crucial role of RCD pathways in the development of sepsis. Notably, ZDHHC3 and TLR5 were identified as novel and robust biomarkers for sepsis.
Background
Sepsis is a life-threatening syndrome that accounts for a substantial proportion of cases among critically ill patients (Vincent et al., 2013). It is frequently accompanied by varying degrees of multiple organ dysfunction, and clinical outcomes may differ significantly due to factors such as geographic disparities, microbial heterogeneity, and individual variability. Despite the advancements in our understanding of the underlying mechanism of sepsis, the mortality rate remains alarmingly high—persistently exceeding 25–30% and reaching up to 40–50% in severe cases (Vincent et al., 2014). These statistics highlight the urgent need to improve the understanding of sepsis.
Regulatory cell death (RCD) is an umbrella term encompassing various forms of programmed cellular events that occur under both physiological and pathological conditions. Various RCD pathways have been identified and extensively studied, including NETotic cell death, oxeiptosis, alkaliptosis, anoikis, among others (Legrand et al., 2019; Del Re et al., 2019). In sepsis, inflammatory episodes and immunodeficiency lead to alterations in intra- and extracellular homeostasis, including immune cell apoptosis, parenchymal cell injury, and dysregulation of organelles such as mitochondria and lysosomes. Thus, RCD is hypothesised to play a critical role in the development of sepsis. Nevertheless, a systematic and comprehensive understanding of the contribution of RCD to sepsis remains lacking.
We aim to identify and characterize a panel of sepsis-associated, RCD-related target genes through multiple analytical approaches. This strategy may provide valuable insights for both basic research and the development of novel therapeutic interventions for sepsis.
Materials and Methods
Animals
Male C57BL/6J (6–8 weeks, 20–25 g) were allowed free access to food and water. The mice were maintained in a standardized housing environment, with temperature and relative humidity controlled at 22 ± 2 °C and 60 ± 5% respectively. The mice were procured from Ziyuan Laboratory Animal Technology Co., Ltd. (Hangzhou, Zhejiang, China). The production qualification of experimental animals was approved by the Zhejiang Provincial Department of Science and Technology (No. SCXK [Zhe] 2024-0004). All experimental procedures were performed in compliance with the guidelines of the Ethics Committee for Laboratory Animals of Anhui Medical University and Centre for Laboratory Animal Research of Anhui Medical University (Approval LLSC-20242423). Mice were randomly assigned and housed in standard cages (30 × 20 × 15 cm).
Cecal ligation and puncture (CLP) surgery
After a one-week acclimatization phase, twelve mice were arbitrarily divided to three groups: a sepsis group with mice sacrificed at 6 and 24 h post-operation, and a sham group. Prior to the procedures, all mice underwent a 6-hour fast with ad libitum access to water. Mice in the sepsis group underwent cecal ligation and puncture (CLP) surgery. Briefly, after anesthesia with 2.0% pentobarbital sodium (50 mg/kg, intraperitoneally), a 2-cm incision was conducted in the abdominal wall. The cecum was then exposed, and feces were gently pushed from the proximal end to the distal end. The cecum was ligated at 75% of its length from the tip using 4-0 silk sutures. A 22-gauge needle was used to puncture the distal cecum, allowing a small quantity of fecal material to be released. The ligated cecum was then returned to the abdominal cavity, and the incision was closed with 4-0 silk sutures. In the sham group, all surgical procedures were conducted except for feces manipulation, ligation, and puncture. Following CLP surgery, mice showed signs of sepsis, including piloerection, diarrhea, and lethargy. Postoperative care and monitoring were implemented to safeguard the health and welfare of the animals, including access to sufficient water and food; efforts were made to minimize pain and discomfort, promoting proper wound healing. At the end of the experiment, mice were anesthetized with an agent that induced rapid unconsciousness to minimize pain and distress. Under light isoflurane anesthesia, blood specimens were collected through retro-orbital venous plexus sampling to reduce discomfort as much as possible. Mice were monitored for any signs of distress following the procedure. Mice that died prematurely were not included in the analysis as they were unable to provide the relevant data. Tissue and blood sample collection was performed by personnel blinded to the group assignments.
Data acquisition
Bulk sequencing datasets (GSE28750, GSE95233, GSE65682, GSE69528, GSE26440, GSE26378, and GSE13904) (Sutherland et al., 2011; Tabone et al., 2019; Scicluna et al., 2015; Pankla et al., 2009; Wong et al., 2009b; Wynn et al., 2011; Wong et al., 2009a) and single-cell RNA sequencing (scRNA-seq) dataset (GSE167363) (Qiu et al., 2021) were all obtained from the GEO database. Regulatory cell death (RCD)-related genes were retrieved from the GeneCards database, the Molecular Signatures Database (MSigDB), and relevant review articles (Zou et al., 2022). RCD genes with relevance scores below the mean level, as determined by the GeneCards database, were excluded based on a filtering criterion. Finally, a list of genes associated with 18 distinct patterns of cell death was compiled (Table S1).
Data processing
The GSE28750 dataset included 20 healthy controls, 10 sepsis patients, and 11 post-surgical participants. The GSE95233 included 51 septic shock patients and 22 healthy controls. After selecting sepsis/septic shock patients and healthy controls, we merged GSE28750 and GSE95233, which share the same GPL platform (GPL570), using the “SVA” algorithm (Leek et al., 2012). Finally, a combined dataset including 61 sepsis/septic shock patients and 42 healthy controls was used as the testing cohort. Differentially expressed genes (DEGs) analysis was performed through the “limma” R package. The datasets GSE65682 (479 sepsis patients and 42 controls), GSE69528 (83 sepsis and 28 controls), GSE26440 (98 sepsis and 32 controls), GSE26378 (82 sepsis and 21 controls), and GSE13904 (158 sepsis and 18 controls) are all considered as validation groups. We used the “Seurat” R package to analyze scRNA-seq data. The analysis workflow included Seurat object creation, quality control, normalization, identification of highly variable genes (HVGs), and dimensionality reduction. Quality control thresholds were defined as follows: (1) the number of unique molecular identifiers (nUMIs) between 500 and 25,000; (2) the expressed genes per cell (nGene) between 200 and 4,000; and (3) the percentage of mitochondrial genes (mitoRatio) <10% per cell. The “SingleR” R package was employed to annotate each cell based on the Human Primary Cell Atlas and Blueprint Encode databases. Data visualization was primarily performed using uniform manifold approximation and projection (UMAP), along with violin plots.
Pathway and functional enrichment analysis
The Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Set Enrichment Analysis (GSEA) analyses were performed using the “clusterProfiler” package to enhance the understanding of gene functions and associated pathways (The Gene Ontology Consortium, 2019; Kanehisa et al., 2016; Yu et al., 2012). Gene Set Variation Analysis (GSVA) was conducted using the “GSVA” package to investigate functional differences in various cell death pathways between septic patients and control samples (Hänzelmann, Castelo & Guinney, 2013).
Immune analysis
For the purpose of immune analysis, CIBERSORT was applied to estimate composition and abundance of immunocytes in sepsis and healthy controls through deconvolution algorithm (Chen et al., 2018).
Construction of the weighted gene co-expression networks (WGCNA) and identification of key modules
Weighted Gene Co-expression Network Analysis (WGCNA) employed to identify the modules most relevant to RCD pathways (Langfelder & Horvath, 2008). To ensure the co-expression network followed a scale-free topology, we calculated the soft-threshold power, and distinct modules were identified via the dynamic tree-cutting method. The module most strongly associated with each RCD pathway was selected for further analysis. Receiver operating characteristic (ROC) analysis was employed to assess how effectively each cell death pathway diagnosed sepsis, dependent on the area under the curve (AUC).
Feature selection of RCD-related genes by various machine learning
Least Absolute Shrinkage and Selection Operator (LASSO) is a regularization method that performs variable selection by shrinking the coefficients of less important variables to zero, thereby reducing model complexity and preventing overfitting, especially in high-dimensional settings. Support Vector Machine (SVM) effectively identifies features that contribute most to classification boundaries by maximizing the margin between classes, making it particularly useful for distinguishing disease from control samples. Random Forest (RF), an ensemble-based algorithm, robustly estimates variable importance by aggregating decision trees and capturing non-linear relationships and variable interactions. To ensure robust and reliable identification of key RCD-associated genes in sepsis, the overlapping genes identified by LASSO, SVM, and RF were considered core RCD-related genes for sepsis, enhancing the stability, generalizability, and biological relevance of the results. The diagnostic efficacy of these hub RCD-related genes was evaluated using ROC analysis. A meta-analysis further confirmed their diagnostic performance. Sensitivity analysis was conducted via the “metainf” function to assess their robustness. Transcription factor (TF) information was obtained from the NetworkAnalyst, while a protein-protein interaction (PPI) network was performed with Cytoscape software (version 3.10.2). As the bulk RNA-seq data were obtained from a public database, only limited clinical metadata (age and sex) were available. To account for potential confounding effects, we performed multivariate Cox regression analyses incorporating gene expression, age, and sex to evaluate survival prognosis. The results were reported as hazard ratios (HRs), 95% confidence intervals (CIs), and p-values.
Causal analysis between ZDHHC3, TLR5 and sepsis-related outcomes
After localizing the expression of Zinc Finger DHHC-Type Containing 3 (ZDHHC3) and Toll-Like Receptor 5 (TLR5) at the single-cell level, a Mendelian randomization (MR) approach was used to further investigate their potential relationship with sepsis. The individuals involved in the MR analysis were of European descent. The cis-expression quantitative trait locus (cis-eQTL) data were collected from the eQTLGen Consortium. The outcome data were derived from the UK Biobank based on the IDs obtained from the Integrative Epidemiology Unit (IEU) OpenGWAS project database. The outcomes analyzed encompassed three parts: the incidence of sepsis, sepsis requiring critical care admission, and 28-day mortality within the intensive care unit (ICU).
The MR analysis was dependent upon three key assumptions: (1) the instrumental variables (IVs) are correlated with the exposure; (2) there are no confounding factors affecting the IV–outcome association; and (3) the IVs influence the outcome solely through the exposure. Initially, IVs with a p-value less than 5 × 10−8 were identified as single nucleotide polymorphisms (SNPs) associated with the exposure, fulfilling assumption 1. Subsequently, linkage disequilibrium (LD) clumping was performed to identify independent SNPs, satisfying assumption 2. Cis-eQTLs were defined as SNPs located within 1 megabase (Mb) of the target gene. In the cis-eQTL analysis, LD was defined as having an r2 < 0.3 within a clumping distance of 100 kilobases (kb). Next, the same SNPs present in the outcome data were extracted. The effect alleles of SNPs were then harmonized to ensure consistency between the exposure and outcome datasets. Palindromic SNPs exhibiting intermediate allele frequencies were excluded to preclude strand ambiguity. Meanwhile, F-statistics were computed to evaluate the strength of instrumental variables (IVs). Finally, MR results were obtained through the “mr” function (TwoSampleMR R package) and were evaluated in terms of odds ratios (ORs). Furthermore, sensitivity analysis was conducted. Heterogeneity and pleiotropy were assessed primarily using MR-Egger and inverse variance weighted (IVW) methods.
Hematoxylin and Eosin (H&E) staining
The lungs of mice were excised and then fixed in 4% paraformaldehyde (PFA) for at least 24 h and embedded in paraffin. The tissue was sectioned into four µm slices for Hematoxylin and Eosin (H&E) staining, and lung pathological injury was evaluated under a light microscope.
Quantitative Real time Polymerase Chain Reaction (qRT-PCR)
The whole blood of each mouse was obtained through orbital blood collection and collected in an Ethylenediaminetetraacetic acid (EDTA)-treated tube. qRT-PCR was utilized to validate the mRNA expression level of the target genes. Total RNA from whole blood was derived using the BIOG Blood RNA Isolated Kit (51025) BIOG, Jiangsu, China) in accordance with the supplier’s protocol. mRNA was reverse-transcribed into complementary DNA (cDNA) using the cDNA Synthesis Kit (A240-10, GenStar, Jiangsu, China). mRNA expression levels were quantified using SYBR premix (A308-05, GenStar, Jiangsu, China) on the CFX Connect™ Real-Time System (Bio-Rad, Hercules, CA, USA). The 20 µl reaction mixture contained two µl cDNA, 10 µl of 2 ×RealStar Universal SYBR qPCR Mix, 0.4 µl each of forward and reverse primers (10 µM), and 7.2 µl of sterile water. The cycling conditions were: 95 °C for 2 min (initial denaturation), followed by 40 cycles of 95 °C for 15 s (denaturation), 60 °C for 30 s (annealing), and 72 °C for 30 s (extension). The relative levels of mRNA were determined via the 2ˆ-ΔΔCT method. Primer sequences were synthesized by Shanghai Shenggong Technology Co., Ltd. and listed in Table S2. The specificity of primers was evaluated through Primer-Basic Local Alignment Search Tool (BLAST) and melting curve analysis.
Statistical analysis
R software packages were used in all statistical analyses (version 4.4.1). The t-test and analysis of variance (ANOVA) were used to compare the means of data following a normal distribution. For data violating the normality assumption, the Kruskal–Wallis test was employed. Correlation coefficients were calculated using Pearson or Spearman correlation analysis. A p-value less than 0.05 was deemed statistically significant.
Results
Identification of RCD-DEGs (RRDs) and enrichment functional analysis
After merging two datasets and correcting for batch effects (Fig. S1), we identified 1,320 DEGs between the sepsis and healthy groups (adjusted p-value < 0.05 and absolute value of log2 fold change (FC) > 0.585). Among them, 501 were upregulated and 819 were downregulated in the sepsis group (Fig. 1A). GSEA showed enrichment in oxidative phosphorylation, xenobiotic metabolism, and mTORC1 signaling, and negative enrichment in interferon-γ response and allograft rejection (Fig. 1B). GO analysis indicated that in the BP category, these genes were primarily involved in leukocyte differentiation, activation, and adhesion. In the cellular component (CC) category, the genes were linked to various lumen-related structures, while in the molecular function (MF) category, they were associated with immune receptor activity (Fig. 1C). KEGG pathway analysis revealed enrichment in the programmed cell death protein 1 (PD-1) checkpoint and nuclear factor-κB (NF-κB) signaling, and the differentiation pathways of Th1, Th2, and Th17 cells (Fig. 1D).
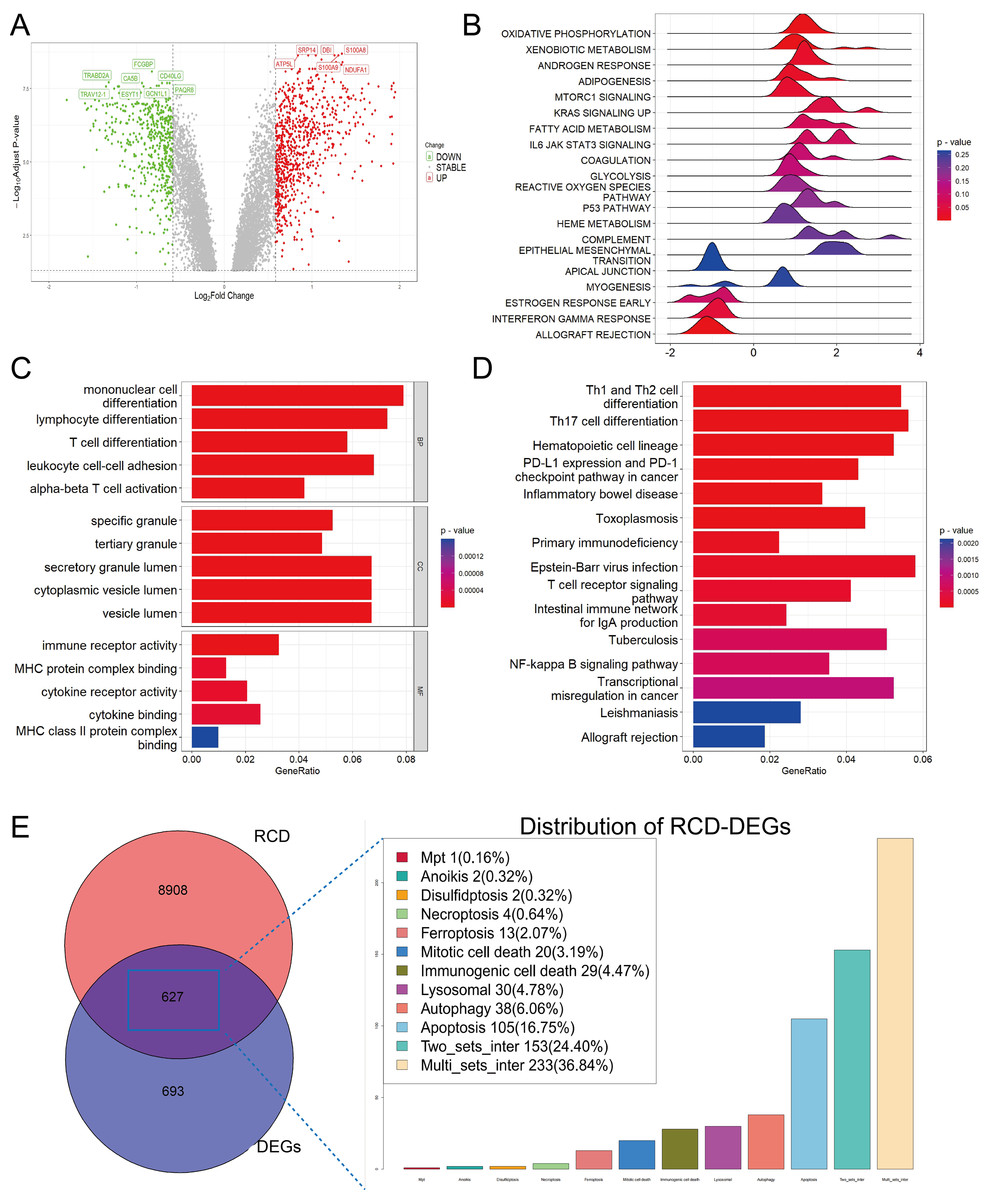
Figure 1: Functional enrichment analysis of DEGs among sepsis and healthy controls.
(A) Volcano plot of DEGs. (B, C, D) Ridge plot and barplot of GSEA, GO, and KEGG enrichment analysis for DEGs. (E) Venn and boxplot of RRDs.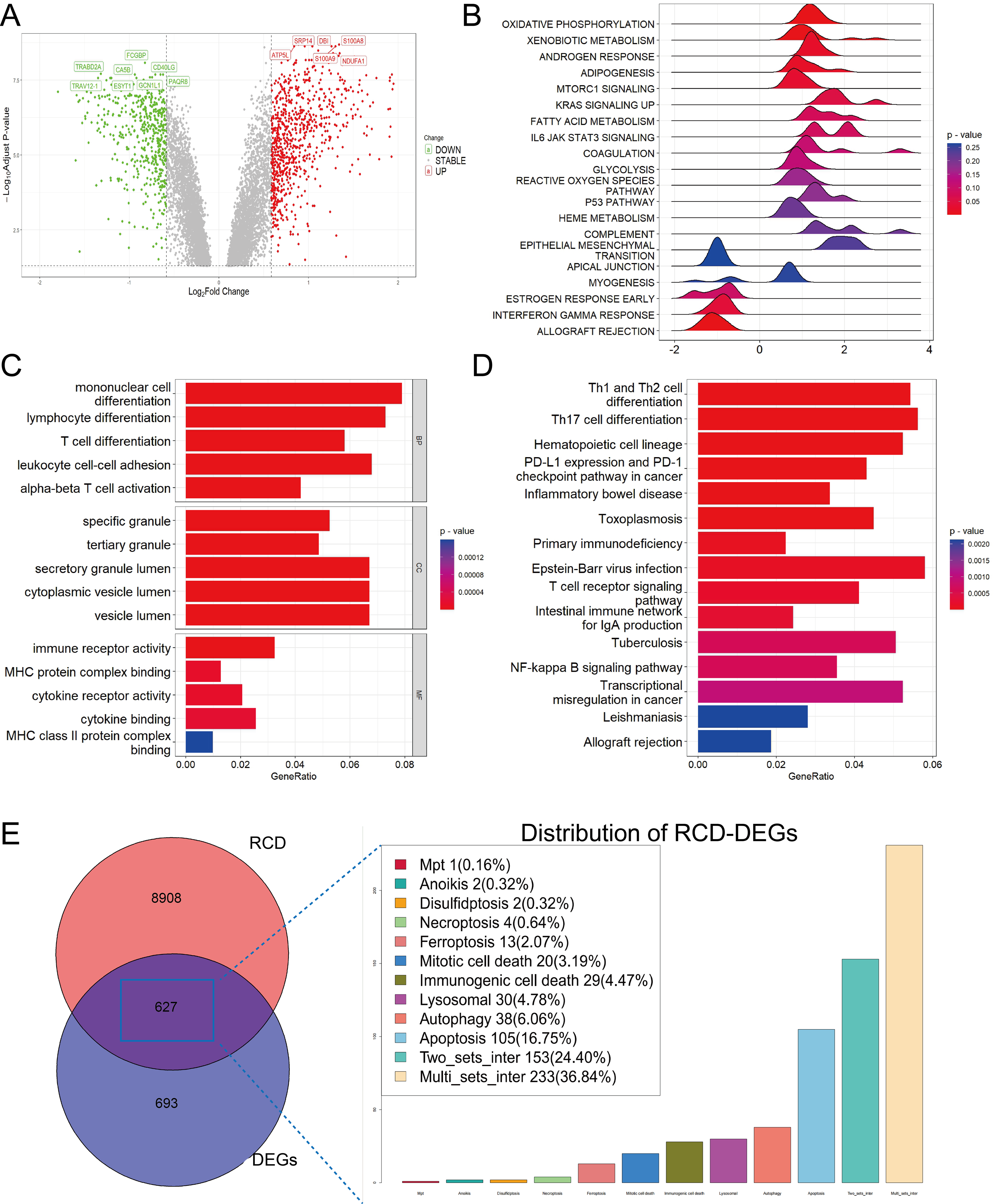{kind=link}
A comprehensive set of RCD-related genes was compiled, and their involvement in multiple biological pathways suggested a complex and systemic role (Fig. S1). Intersecting these genes with DEGs yielded 627 RRDs (Fig. 1E), which were subjected to functional analyses (Fig. S2). GSEA revealed a positive enrichment in xenobiotic metabolism, mTORC1 signaling, coagulation, and IL6-JAK-STAT3 signaling, and negative enrichment of inflammatory response, interferon-γ response, and allograft rejection. GO and KEGG analyses yielded results consistent with the previous enrichment analysis of DEGs.
Characterization of hub RCD pathways and RRDs
CIBERSORT analysis revealed higher levels of immune cell infiltration in sepsis compared to healthy controls. GSVA analysis was utilized to assess the enrichment scores of each RCD pathway for every sample. Septic patients showed higher enrichment of NETotic cell death, oxeiptosis, alkaliptosis, anoikis, disulfidptosis, lysosomal cell death, necroptosis, pyroptosis, mitochondrial permeability transition (MPT), ferroptosis, immunologic cell death, apoptosis, and lower enrichment in cuproptosis, parthanatos, autosis, and autophagy (Fig. S1). Except for parthanatos, autosis, necroptosis, autophagy, and entotic cell death, all other RCD pathways demonstrated statistically significant correlation with immune cells in sepsis (Fig. 2A). Subsequently, we used one-step network construction function of the WGCNA R package to identify gene modules strongly associated with oxeiptosis, lysosomal cell death, ferroptosis, anoikis, and pyroptosis, respectively (Fig. 2B). The diagnostic values (AUCs) for each pathway were 0.8989, 0.8646, 0.9212, 0.8314, and 0.8177, respectively (Fig. 2C). In conclusion, oxeiptosis, lysosomal cell death, ferroptosis, anoikis, and pyroptosis were identified as key RCD pathways in sepsis.
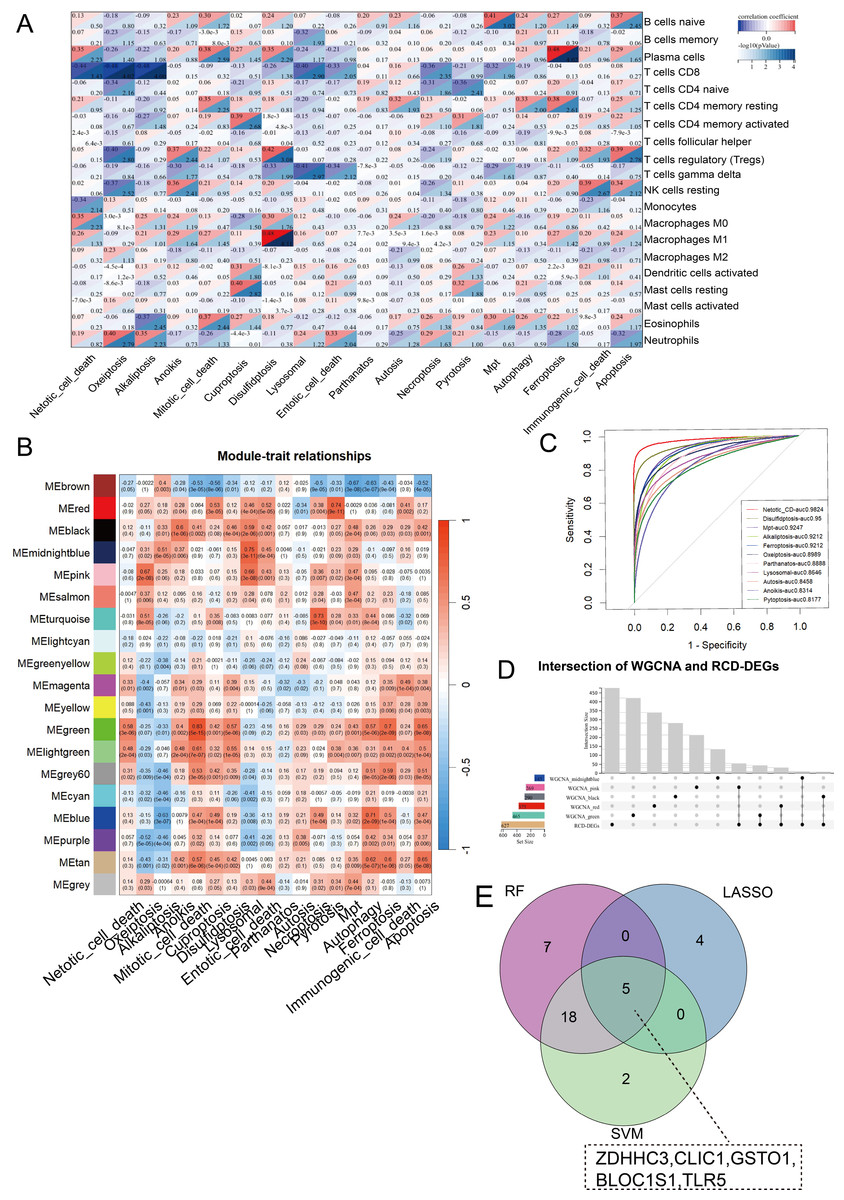
Figure 2: Characterization of hub RCD pathways and RRDs.
(A) Correlation analysis of immune infiltrating and RCD pathway enrichment scores. (B) Heatmap of the correlations between module genes and 18 RCD pathways. (C) ROC analysis of each RCD in septic diagnosis. (D) Upset plot of intersection of module genes and DEGs of sepsis. (E) Venn diagram of the intersection of LASSO, SVM, RF.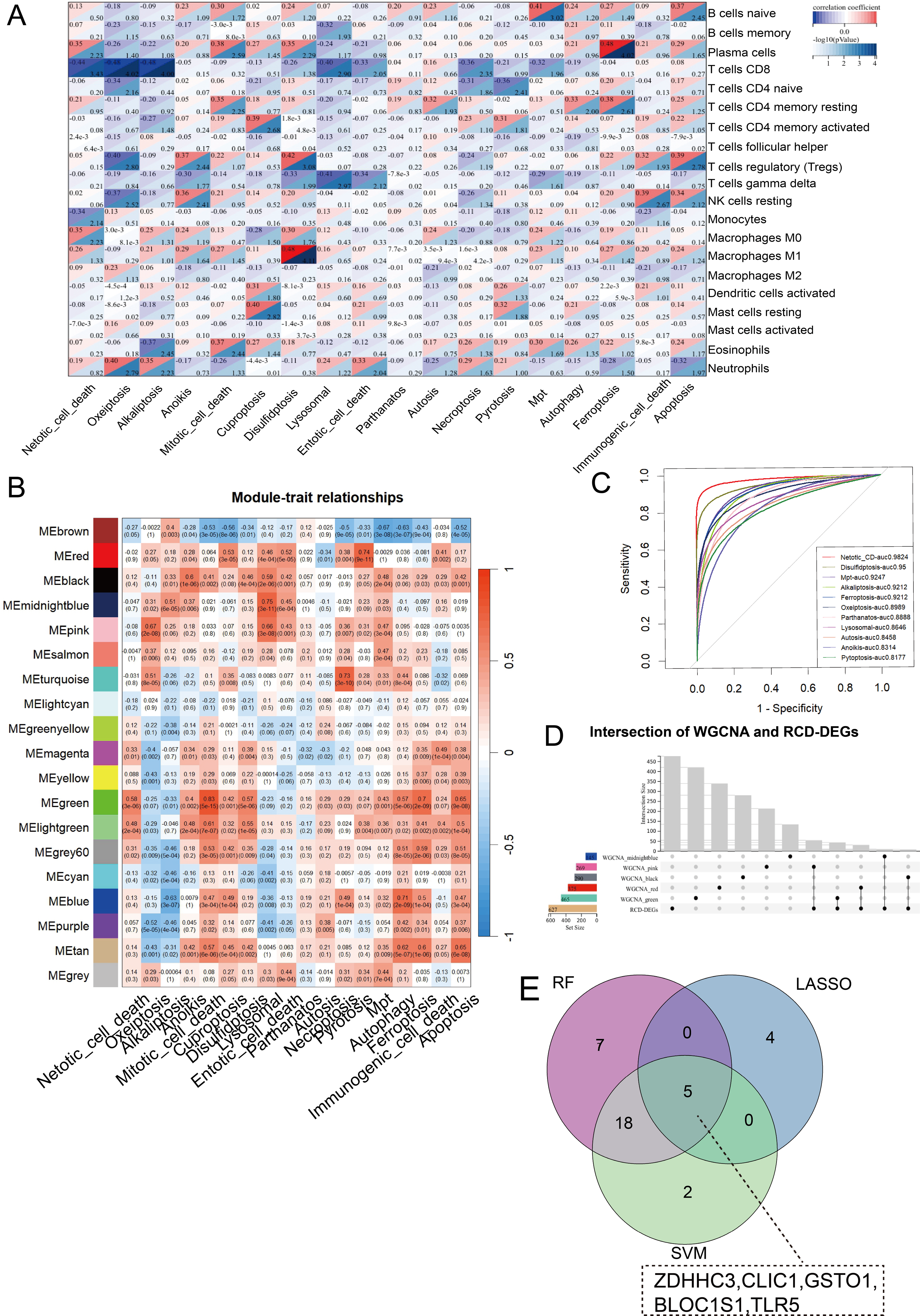{kind=link}
By integrating WGCNA results, we identified an additional 151 RRDs (Fig. 2D). GO analysis of these 151 genes exhibited enrichment in innate immune activation, response to bacteria and lipopolysaccharide, molecules of bacterial origin, and cell–cell adhesion, while KEGG analysis revealed enrichment in the nucleotide-binding oligomerization domain (NOD)-like receptor signaling pathway, cellular senescence, and neutrophil extracellular trap formation (Fig. S2). These 151 genes were further evaluated using LASSO, RF, and SVM algorithms to ensure robust feature selection (Fig. S3). Each algorithm offers unique advantages in identifying informative features. By focusing on the overlapping genes selected by all three methods, we aimed to reduce model-specific bias and enhance the biological relevance and reproducibility of identified biomarkers. Consequently, five hub RRDs were identified: ZDHHC3, Chloride Intracellular Channel 1 (CLIC1), Glutathione S-Transferase Omega 1 (GSTO1), Biogenesis of Lysosomal Organelles Complex 1 Subunit 1 (BLOC1S1), and TLR5 (Fig. 2E).
Immune localization analysis for hub RRDs at single-cell level
At single-cell level, we annotated 30,192 cells as CD4+ T cells, CD8+ T cells, B cells, erythrocytes, monocytes, neutrophils, natural killer (NK) cells, and platelets (Fig. 3A). The five hub RRDs demonstrated significantly higher expression levels in monocytes and neutrophils (Figs. 3B–3D). Despite their elevated levels in septic group at the bulk level, a diverse distribution was observed at the single-cell level. The bulk-level expression difference of ZDHHC3, GSTO1 and TLR5, primarily originated from monocytes, and BLOC1S1 was derived from both monocytes and neutrophils. In contrast, the expression pattern of CLIC1 appeared to be more complex (Fig. 3E). Furthermore, monocytes and neutrophils from non-survivors showed higher expression levels of ZDHHC3, CLIC1, and GSTO1. Interestingly, BLOC1S1 expression was elevated only in neutrophils in the non-survivor group, while it was significantly lower in monocytes, NK cells, B cells, CD4+ T cells, and CD8+ T cells (Fig. 3E). Finally, cells co-expressing all five hub RRDs were identified based on their median expression cut-offs. The proportion of monocytes co-expressing hub RRDs was significantly higher in sepsis patients, and co-expressing neutrophils were more abundant in the non-survivors (Fig. 3F). Conclusively, monocyte- and neutrophil-related biological processes are likely central to the function of these five hub RRDs.
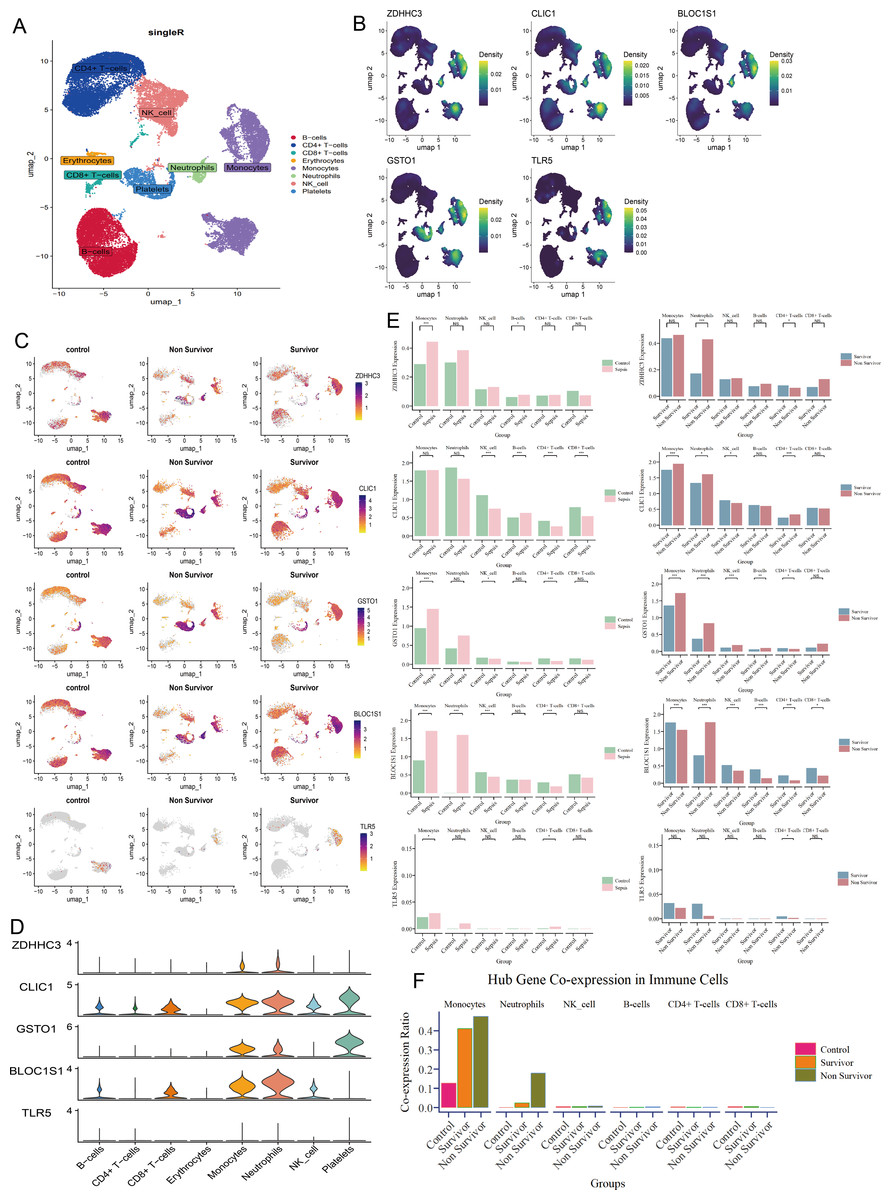
Figure 3: Immune localization analysis for hub RRDs at a single-cell level.
(A) The single cell data were annotated into 8 cell clusters. (B) Density plot of hub RRDs expression. (C) Distribution of hub RRDs among different group. (D) Violin plot of hub RRDs expression. (E) Differential expression analysis for five hub RRDs between control and sepsis group, and between survivor and non-survivor groups.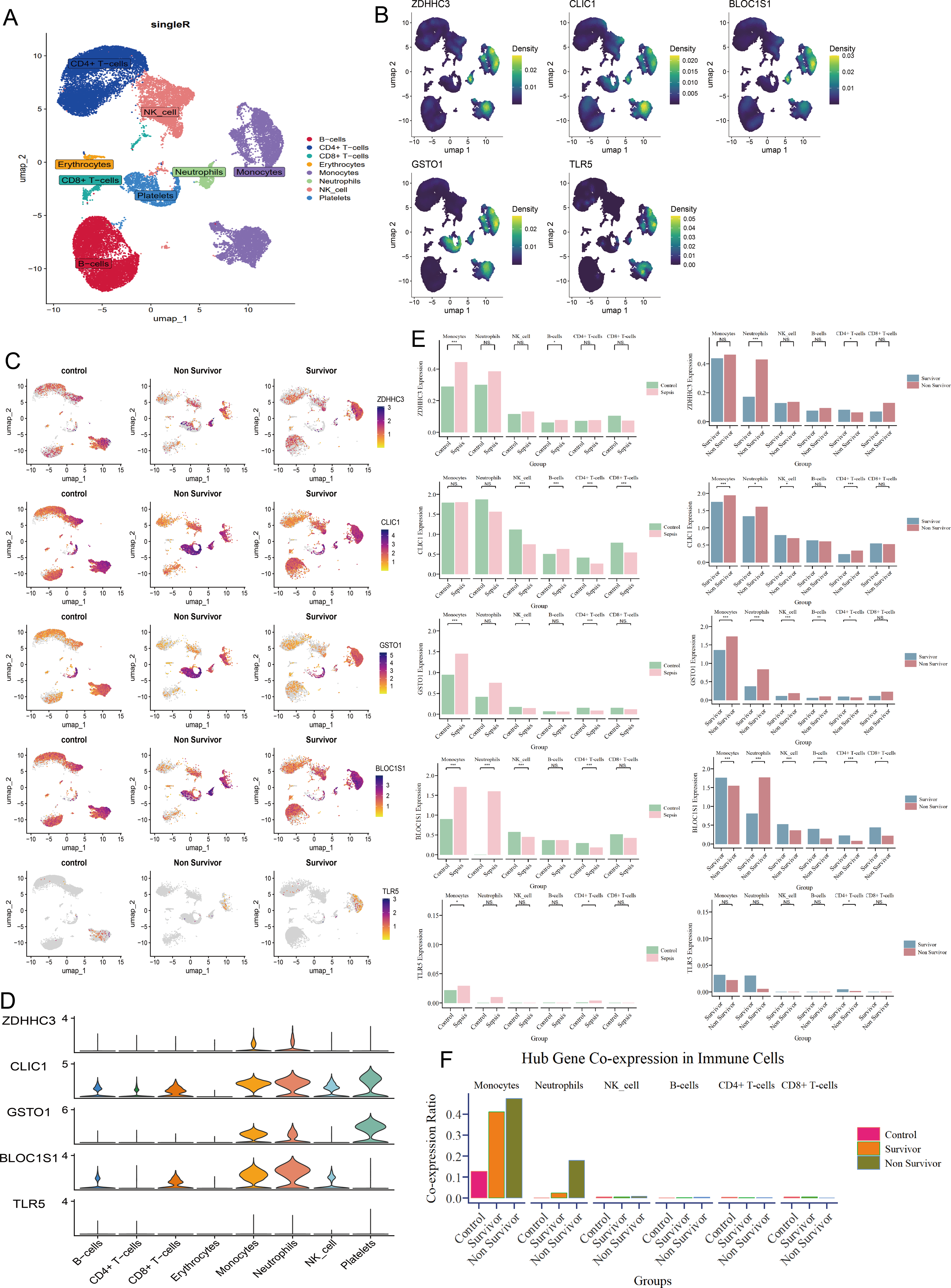{kind=link}
Validation of hub RRDs and their GSEA, PPI, immune correlation analysis
The five hub RRDs identified above demonstrated divergent expression levels between sepsis patients and controls across all validation cohorts. All five genes exhibited strong diagnostic performance, with AUC scores exceeding 0.8 (Figs. 4A–4E). To validate these findings, a meta-analysis using a random-effects model confirmed the diagnostic efficacy of these genes (Fig. S4).
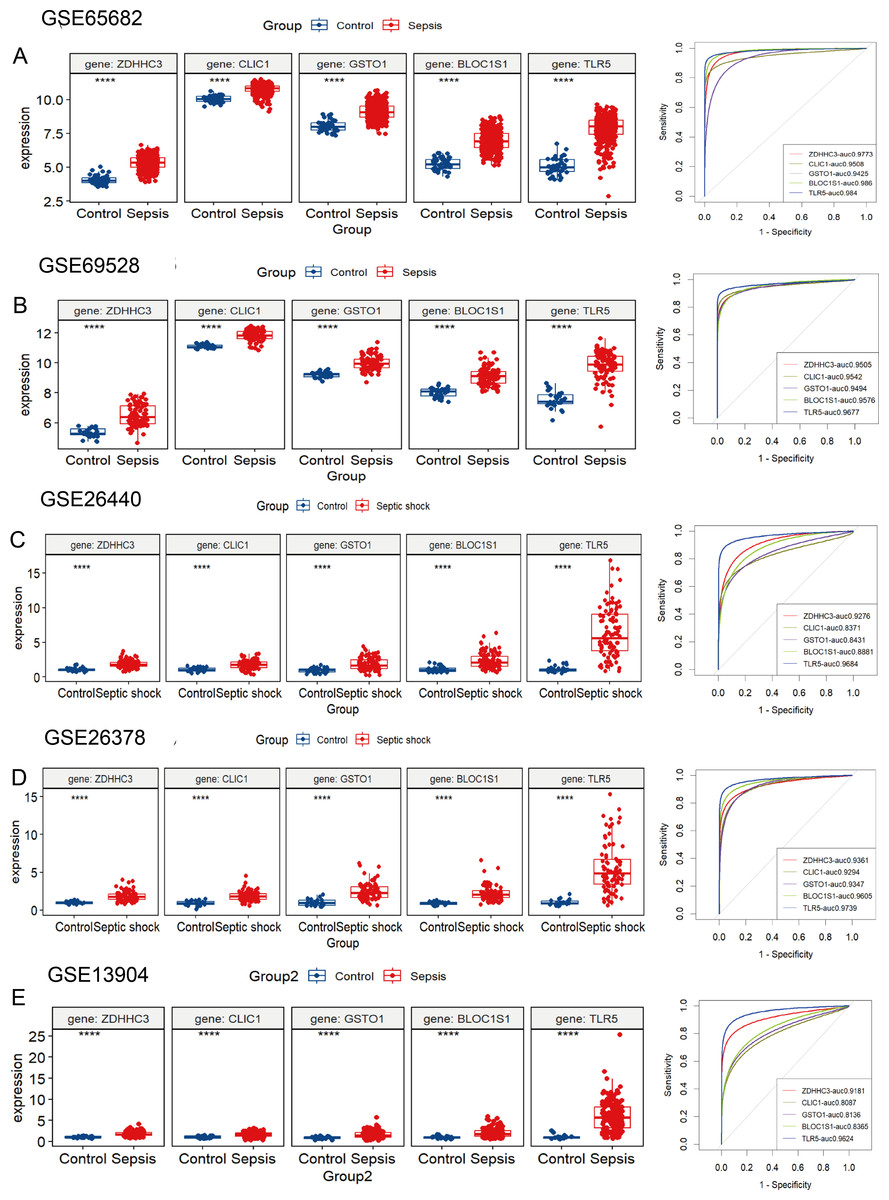
Figure 4: Validation of these five hub RRDs in various cohorts.
(A–E) Differential expression and ROC curve analysis of the five hub RRDs across five independent datasets: GSE65682, GSE69528, GSE26440, GSE26378, and GSE13904.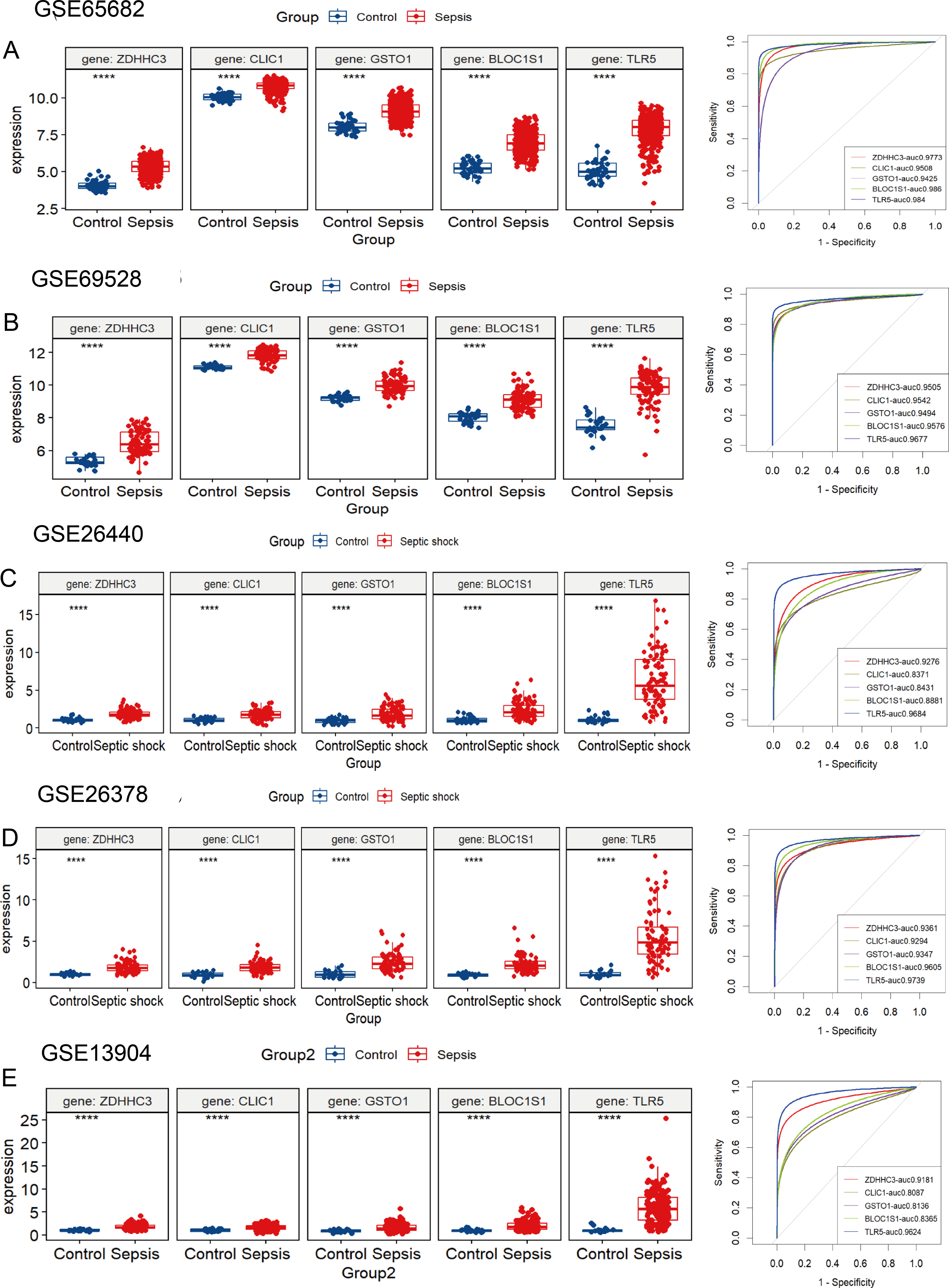{kind=link}
To identify the pathways related to hub RRDs, septic patients were classified into two groups according to the expression levels of the five hub genes, and GSEA was performed (Fig. 5). In addition to previously identified pathways, G2M checkpoint, MYC targets, E2F targets, mTORC1 signaling, PI3K-AKT-mTOR signaling, oxidative phosphorylation, mitotic spindle, protein secretion, and androgen response were activated in sepsis patients with high RRD expression. Meanwhile, pathways related to E2F, G2M, and MYC targets, IFN-γ/α responses, and inflammatory responses were inhibited in the high-RRD group. Additionally, pathways involving DNA repair, heme metabolism, Kirsten rat sarcoma viral oncogene homolog (KRAS) signaling, myogenesis, and allograft rejection were inhibited. Correlation analysis between hub RRDs and immune infiltration scores was also performed. As exhibited in Fig. 6A, ZDHHC3 expression was strongly positively associated with naïve B cells. CLIC1 expression was positively correlated with neutrophils and negatively correlated with CD8+ T cells, similar to the pattern observed for TLR5. GSTO1 expression was negatively correlated with γδ T cells. The PPI network analysis indicated that FOXC1 may serve as a key TF interacting with all five hub genes (Fig. 6B). To further evaluate the prognostic value of the hub RRDs, multivariate Cox analysis was performed. The results revealed that age (HR: 1.02; 95% CI [1.01–1.03]; p < 0.05), ZDHHC3 (HR: 1.74; 95% CI [1.19–2.54]; p < 0.05) and TLR5 (HR: 0.78; 95% CI [0.66–0.92]; p < 0.05) were independent prognostic factors for sepsis survival.
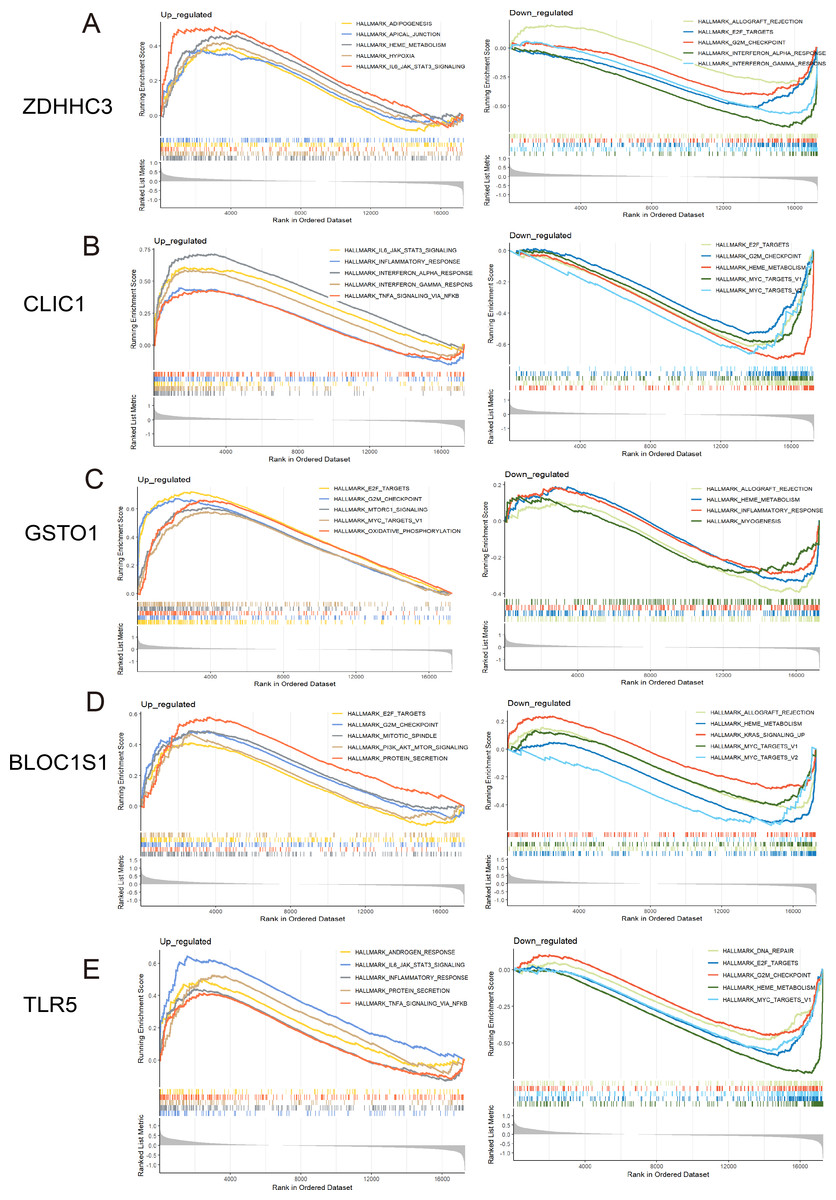
Figure 5: GSEA for the five hub RRDs.
GSEA between distinct groups characterized by different expression levels of ZDHHC3 (A), CLIC1 (B), GSTO1 (C), BLOC1S1 (D), and TLR5 (E).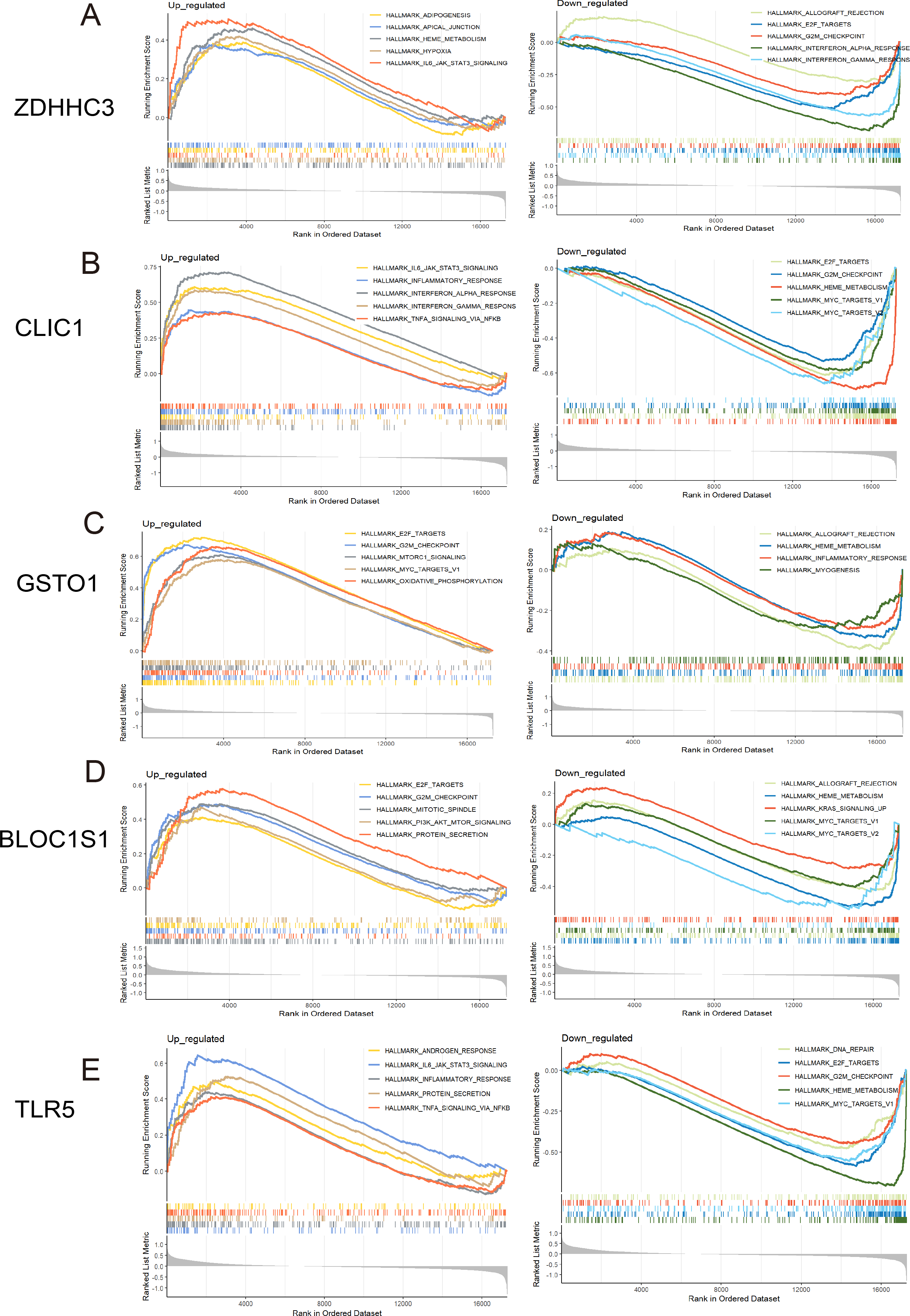{kind=link}
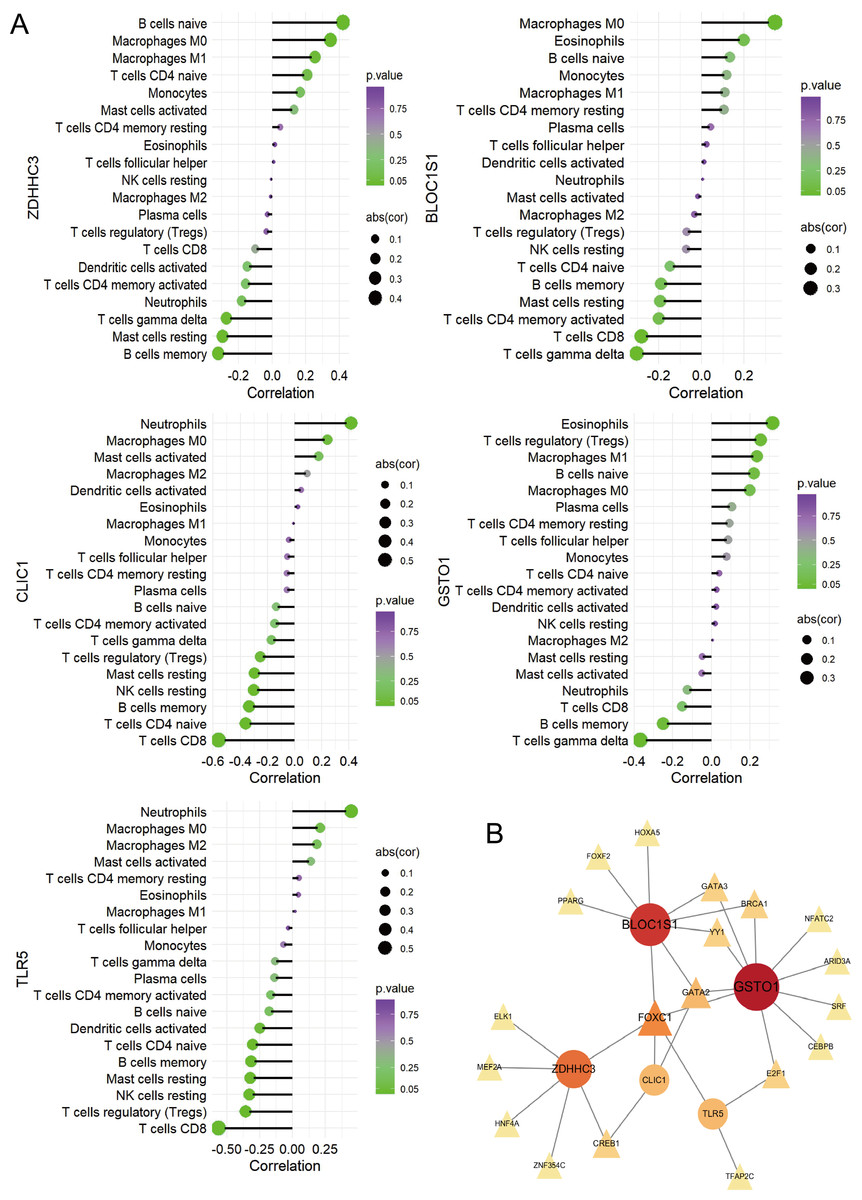
Figure 6: Immune correlation analysis for five hub RRDs.
(A) Immune correlation analysis. (B) PPI analysis.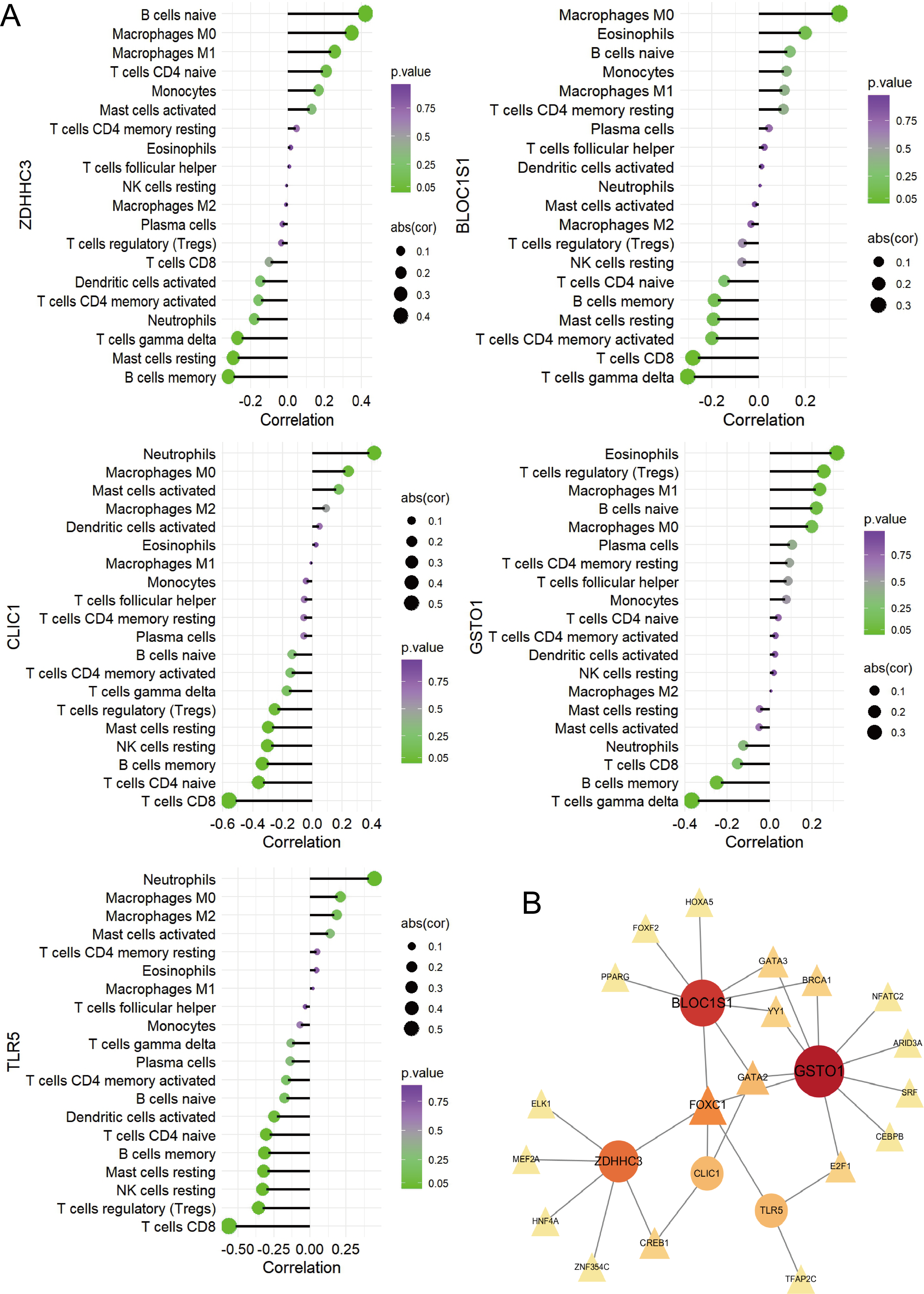{kind=link}
Causal analysis between ZDHHC3, TLR5 and septic related outcome
To evaluate the prognostic value of ZDHHC3 and TLR5 in sepsis, we conducted MR analysis to explore their potential causal roles in sepsis development.
MR analysis showed that the cis-eQTL of ZDHHC3 was associated with an increased risk of ICU admission in sepsis patients, suggesting a potential causal relationship (OR = 1.626; 95% CI [1.115–2.371]; p = 0.012). A sensitivity analysis was then conducted to verify the robustness of the outcomes. MR-Egger (Cochran’s Q = 5.099; p = 0.826) and inverse variance weighted (IVW) methods (Cochran’s Q = 7.285; p = 0.698) were used to assess the heterogeneity. The results indicated no significant heterogeneity. The horizontal pleiotropy test revealed no evidence of pleiotropy among the selected IVs (intercept = −0.080; standard error (SE) = 0.054; p = 0.173). The cis-eQTL of TLR5 was associated with an increased risk of sepsis incidence (OR = 1.136; 95% CI [1.083–1.192]; p = 1.81 × 10−7) and decreased 28-day mortality (OR = 0.65; 95% CI [0.493–0.857]; p = 0.002). These sensitivity analyses further confirmed the stability of the findings. The p-values for heterogeneity and pleiotropy tests were all above 0.05, indicating no significant bias.
Validation in septic model
Considering the independent significance of ZDHHC3 and TLR5 in predicting septic survival and their potential causal relationships with sepsis development, we examined the levels of Zdhhc3 and Tlr5 in a septic mouse model.
Significant neutrophil infiltration and alveolar edema were observed in the CLP group (Fig. 7A). Subsequently, qRT-PCR was utilized to quantify the expression levels of Zdhhc3 and Tlr5 in each sample. The results showed that the Zdhhc3 expression was increased in the septic group at 6 h post-induction, but significantly decreased by 24 h (p < 0.0001). A slight decline was observed in the 24-hour group compared to the sham group, but it was not statistically significant (Fig. 7B). A similar expression pattern was observed for Tlr5 (Fig. 7C). Consistently, both Zdhhc3 and Tlr5 exhibited increased expression at the 6-hour time point.
Discussion
Our research successfully identified five key immune-related RRDs—ZDHHC3, CLIC1, GSTO1, BLOC1S1, and TLR5—by integrating multiple machine learning methods. Their pathological relevance to monocytes and neutrophils was also emphasized. To explore the independent predictive value and potential causal relationships of ZDHHC3 and TLR5 in sepsis, we validated their expression levels in a septic mouse model and confirmed their significant upregulation at 6 h after sepsis onset.
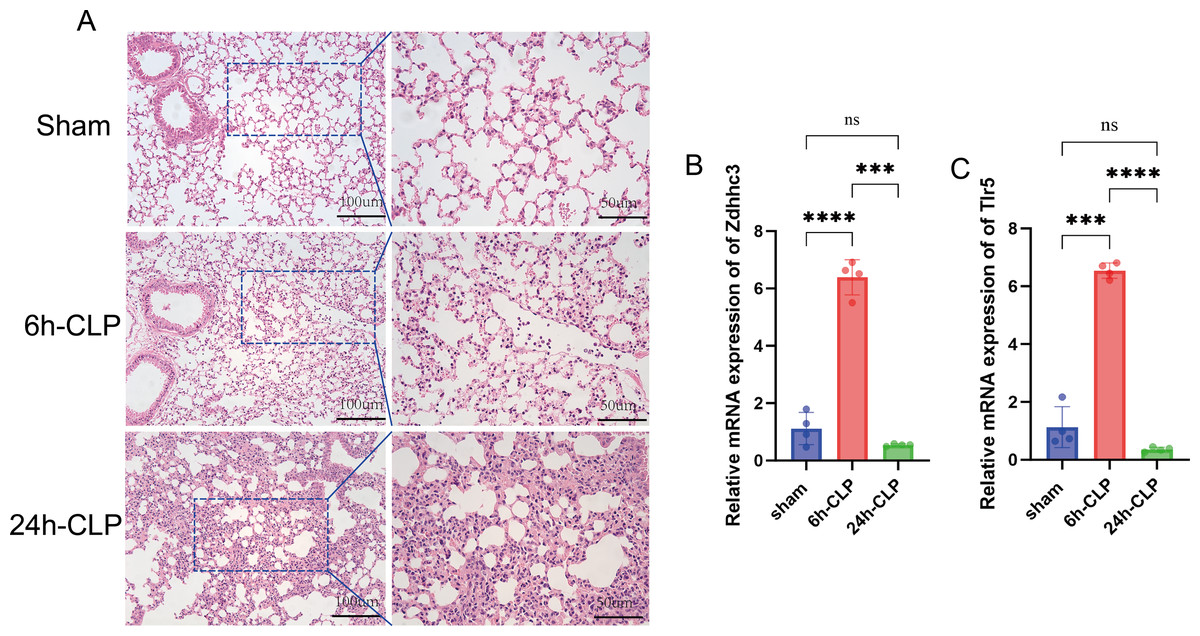
Figure 7: Pathological changes of septic lungs in mice and the results of qRT-PCR experiment.
(A) Results of H&E staining of lungs. (B) The expression level of Zdhhc3. (C) The expression level of Tlr5. Data are presented as the mean ± SD. ****p < 0.0001. ***p < 0.001.{kind=link}
Varying enrichment levels of RCD, their correlations with immune cells, and strong diagnostic efficacy collectively exhibit the complex and critical roles of cell death patterns in sepsis. Oxeiptosis, lysosomal cell death, ferroptosis, anoikis, and pyroptosis were identified as key RCD pathways in our research. Oxeiptosis, first conceptualized in 2018, is a pattern of cell death marked by reactive oxygen species (ROS) sensitivity, caspase independence, and a non-inflammatory mechanism that helps protect tissues from ROS-induced inflammation (Holze et al., 2018). In Oikawa et al. (2022) confirmed that the deubiquitinase OTUD1 regulates KEAP1-mediated oxeiptosis in a sepsis model. However, research on oxeiptosis in sepsis remains limited, highlighting promising directions for further investigation. Lysosomal cell death is characterized by lysosomal rupture, mediated by protease or iron. This process may be further intensified and complicated in the presence of apoptosis, ferroptosis, and autophagy. Recent studies have shown that lipopolysaccharide (LPS)-induced ROS can promote lysosomal cell death in vitro, offering insights into the mechanisms underlying liver dysfunction in septic patients (Hsu et al., 2024). However, further research in this area is still lacking. A surge in inflammatory cytokine production is a hallmark of early-stage sepsis. Mechanistically, pyroptosis is a key driver of inflammation during sepsis. It has been extensively discussed in the initiation and progression of sepsis, with evidence showing that targeting pyroptosis in macrophages, neutrophils, and other parenchymal cells may help alleviate sepsis-induced injury (Zhao et al., 2024; Jing et al., 2022; Liu et al., 2020). Although ferroptosis is known to be driven by iron accumulation and lipid peroxidation, no specific clinical markers have yet been identified for sepsis patients. Notably, ZDHHC3 and TLR5 were identified as survival-related genes in our research.
ZDHHC3 was identified as a key member of the most significant anoikis-related module in the present study. Anoikis, a uniqe form of apoptosis, is triggered by loss of cell adhesions to the extracellular matrix or abnormal cell adhesion, and has been widely studied in cancer research (Taddei et al., 2012). Notably, resistance to anoikis is considered as a critical contributor to metastasis and the survival for malignant cells (Shi et al., 2024). Certain oncoviruses can also facilitate this process (Matarrese et al., 2000; Kakavandi et al., 2018). In contrast, anoikis showed significantly upregulated enrichment levels in sepsis, associated with regulatory T cells (Tregs) and resting NK cells. However, studies on anoikis in sepsis, as well as the role of ZDHHC3 in this process, remain limited. ZDHHC3 is a key zinc finger-aspartate-histidine-histidine-cysteine (DHHC)-cysteine rich domain (CRD)-type palmitoyltransferase that catalyzes palmitoylation and regulates intercellular signaling, protein localization, and function. It plays dual roles in various diseases, including immune responses, neurological disorders, and cancer (Spinelli et al., 2017; Yao et al., 2019; Lin et al., 2021). Previous studies have shown that elevated ZDHHC3 levels may promote cognition impairment related to metabolic disorders, particularly under a high-fat diet (Lin et al., 2021). Additionally, ZDHHC3-mediated palmitoylation of PD-L1 has been linked to T-cell responses in tumors and positively correlated with tumorigenesis (Zhang et al., 2020; Ning et al., 2021). Few studies have explored its mechanisms in the initiation and progression of sepsis. Notably, a recent study showed that ZDHHC3 stabilizes IRHOM2 via S-palmitoylation, preventing its ubiquitin-dependent degradation and prolonging MAP3K7-JNK-NF-κB pathway activation (Xu et al., 2023). The increased level of Zdhhc3 at 6 h in our study may have partially contributed to the hyperinflammatory state observed in sepsis. Interestingly, its homolog, ZDHHC7, has recently been highlighted as a key regulator of NLRP3 activation in macrophages and a potential therapeutic target for various inflammatory diseases (Yu et al., 2024). Other palmitoylating enzymes have also been studied for their roles in pathogen sensing, NLRP3 inflammasome activation, cytokine production, and pyroptosis. Consistently, our findings indicate that ZDHHC3 is independently associated with survival prediction, sepsis incidence, and disease progression. Its elevated expression in monocytes and neutrophils, and increased levels at 6 h, suggest a promising role in sepsis pathophysiology. However, the detailed mechanisms of ZDHHC3-induced anoikis in sepsis remain unclear, particularly its regulation of the NF-κB pathway, NLRP3 assembly, Gasdermin E (GSDME) activation, neutrophil extracellular trap (NET) formation, immune cell recruitment, and oxidative stress during disease onset and progression. Additionally, ZDHHC3 is involved in lysosomal cell death and apoptosis, although most reports have focused on its anti-cancer roles (Kakavandi et al., 2018; Yao et al., 2019). Furthermore, Zdhhc3 also shows clinical therapeutic potential in some research. For example, inhibition of Gasdermin E (GSDME) palmitoylation by 2-BP can reduce chemotherapy-induced cell pyroptosis (Hu et al., 2020). 2-BP can also alter gut microbiota composition and exacerbate endothelial injury. However, the mechanisms of 2-BP in sepsis remain unexplored (Ma et al., 2024; He et al., 2025). Our study is the first to propose the potential link between ZDHHC3 and sepsis progression. However, the underlying connection between ZDHHC3 and sepsis requires systematic and comprehensive investigation.
TLR5, a member of toll-like receptor family, primarily recognizes pathogen-associated molecular patterns (PAMPs), particularly flagellin, leading to activation of the NF-κB pathway (Harrison et al., 2008). Unlike TLR4 which recognizes lipopolysaccharide and is a well-established pathological factor in sepsis, triggering widespread systemic injury, early researches proposed a protective role of TLR5 in initiating immune response. However, these studies did not differentiate between survivors and non-survivors (Silva et al., 2014). In our study, the TLR5 gene was linked to the oxeiptosis-related module. However, the role of TLR5-mediated ROS-associated cell death in sepsis has not been fully investigated. In addition to oxeiptosis, TLR5 has also been linked to apoptosis. Apoptosis remains a major focus of sepsis. Increased apoptosis in macrophages, neutrophils, cardiomyocytes, and renal cells has been linked to heightened inflammation, immune cells depletion, and multiple organs dysfunctions (Ge et al., 2021; Zhang et al., 2019; Wang et al., 2021). Although TLR5-related apoptosis in sepsis has not been extensively discussed, evidence suggests that TLR5 depletion can inhibit hyperammonaemia-induced liver cell apoptosis by suppressing NF-κB pathway and mitogen-activated protein kinase (MAPK) signaling, indicating its dual regulatory effects on sepsis (Yan et al., 2019). Additionally, several studies have identified TLR5 as a robust and influential hub gene linking COVID-19, sepsis, and ARDS (Li et al., 2023). Another research also suggested its diagnostics potential in sepsis (Yang et al., 2022). Interestingly, a 2025 study reported a correlation between TLR5 and up-regulated PD-1/PD-L1 in lung sepsis, implying that interactions between TLRs and these immune checkpoints may contribute to histopathological changes in lung tissue (Sinos et al., 2025). However, the underlying mechanisms behind these interactions were not further explored. In our study, increased TLR5 expression in monocytes and neutrophils may suggest its role in enhancing their recruitment and activation, contributing to inflammatory injury in sepsis. This may heighten innate immune sensing and drive neutrophil-mediated systemic inflammation, consistent with previous studies. qRT-PCR results also showed elevated TLR5 expression at the 6-hour time point. This may indicate early immunological dynamics in sepsis. Further systematic studies are needed to confirm these findings.
By integrating multiple datasets and applying various machine learning techniques, we identified the key biomarkers associated with sepsis. However, several limitations remain in our study. First, the lack of comprehensive survival and clinical data—such as comorbidities, medications, and chronic diseases—limited the validation of these core RRDs. More comprehensive patient data are needed to further evaluate the validity of these RRDs and control for confounding bias. Second, no additional experiments were conducted to investigate the relationships between ZDHHC3, TLR5 and sepsis or sepsis-induced multiple organ dysfunction. Third, the mutual effects of ZDHHC3 and TLR5, as well as their associated molecular and immune cell subtypes, were not further investigated in the present study.
Conclusion
In summary, this study identified five key RRDs that play an instrumental role in the diagnosis of sepsis. Among them, ZDHHC3 and TLR5 were identified as predictors of sepsis survival. The expression of ZDHHC3 and TLR5 was predominantly detected in monocytes and neutrophils, and was significantly elevated in a septic mouse model at 6 h.
Supplemental Information
Identification of DEGs, genes of 18 RCD genes, CIBERSORT, and GSVA analysis of RCD
(A, B) Distribution of samples before and after batch effects removal. (C) Upset of genes of 18 types of RCD. (D) Violin plot of results for CIBERSORT analysis. (E) B oxplot of results for GSVA scores of 18 RCD pathways.
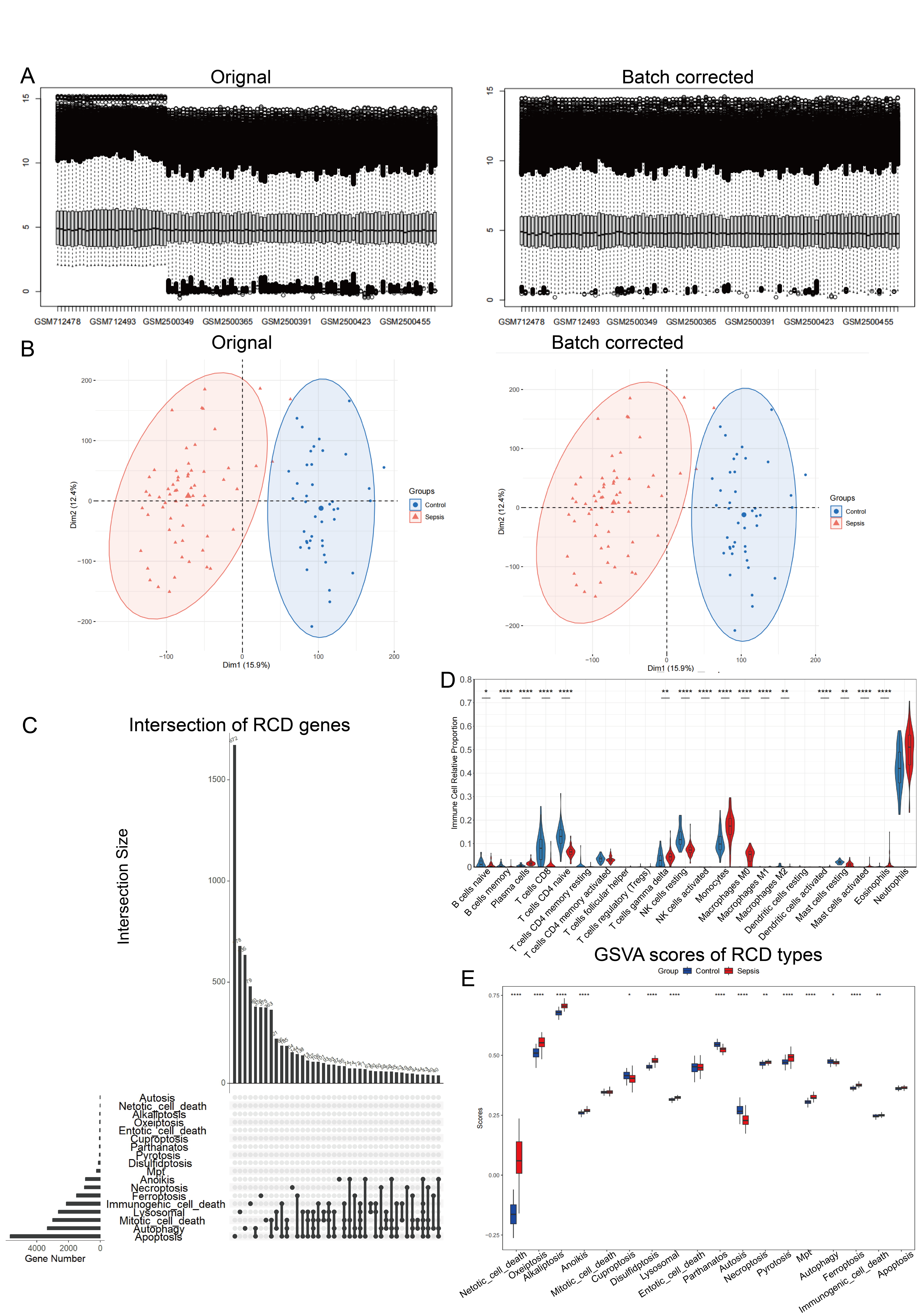{kind=link}
Enrichment analysis of RRDs
(A) GO enrichment analysis of R RD s. (B) KEGG enrichment analysis of R RD s. (C) GSEA enrichment analysis of R RD s.
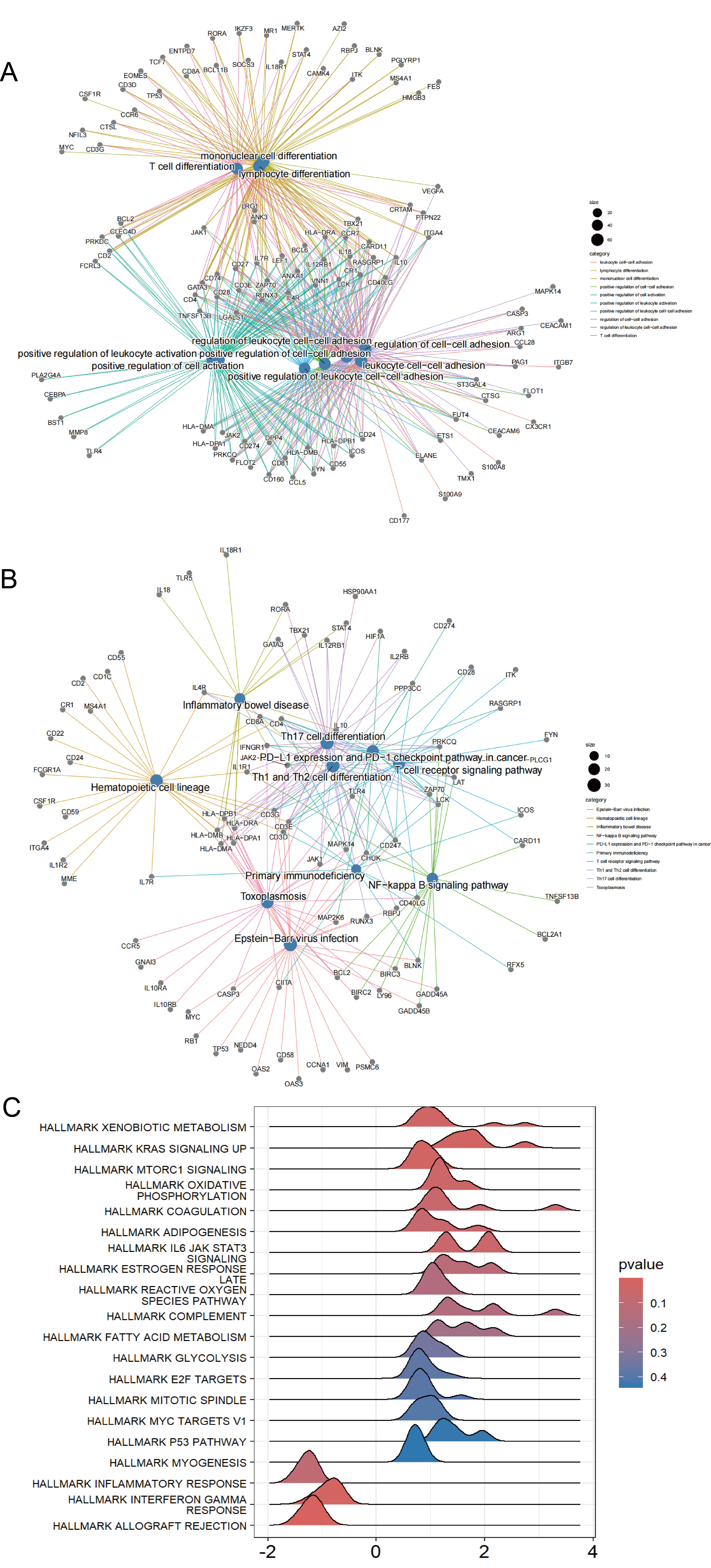{kind=link}
Identification of key RRDs
(A) Distribution of LASSO coefficients for differential genes and ten-fold cross-validation for tuning parameter selection in LASSO model. (B) Distribution of root mean square error (RMSE) in SVM model. (C) Top 30 selected variations in RF model.
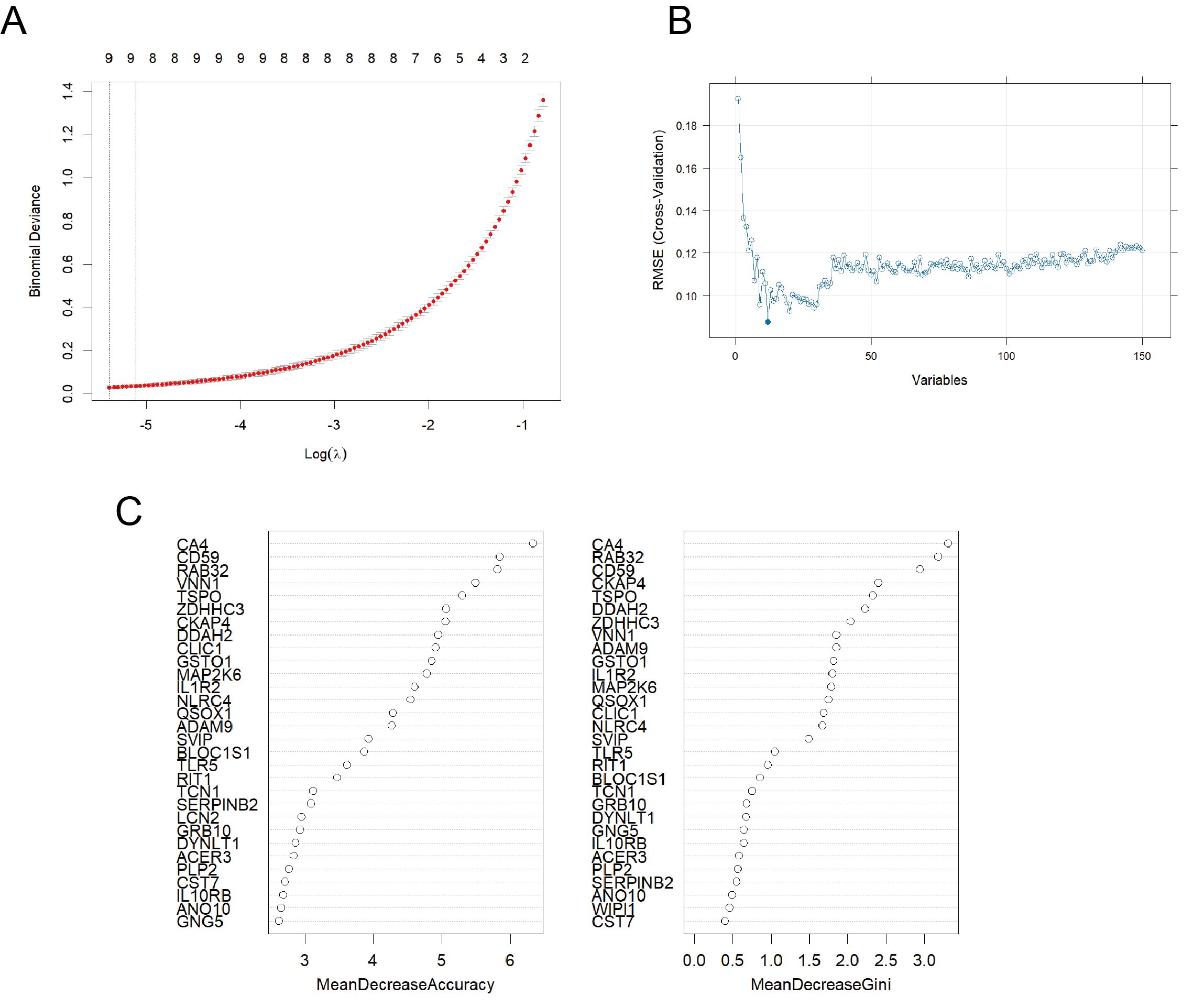{kind=link}
Meta analysis of the differential distribution of the five genes among sepsis and controls
Meta analysis of the differential distribution of the five genes among sepsis and controls.
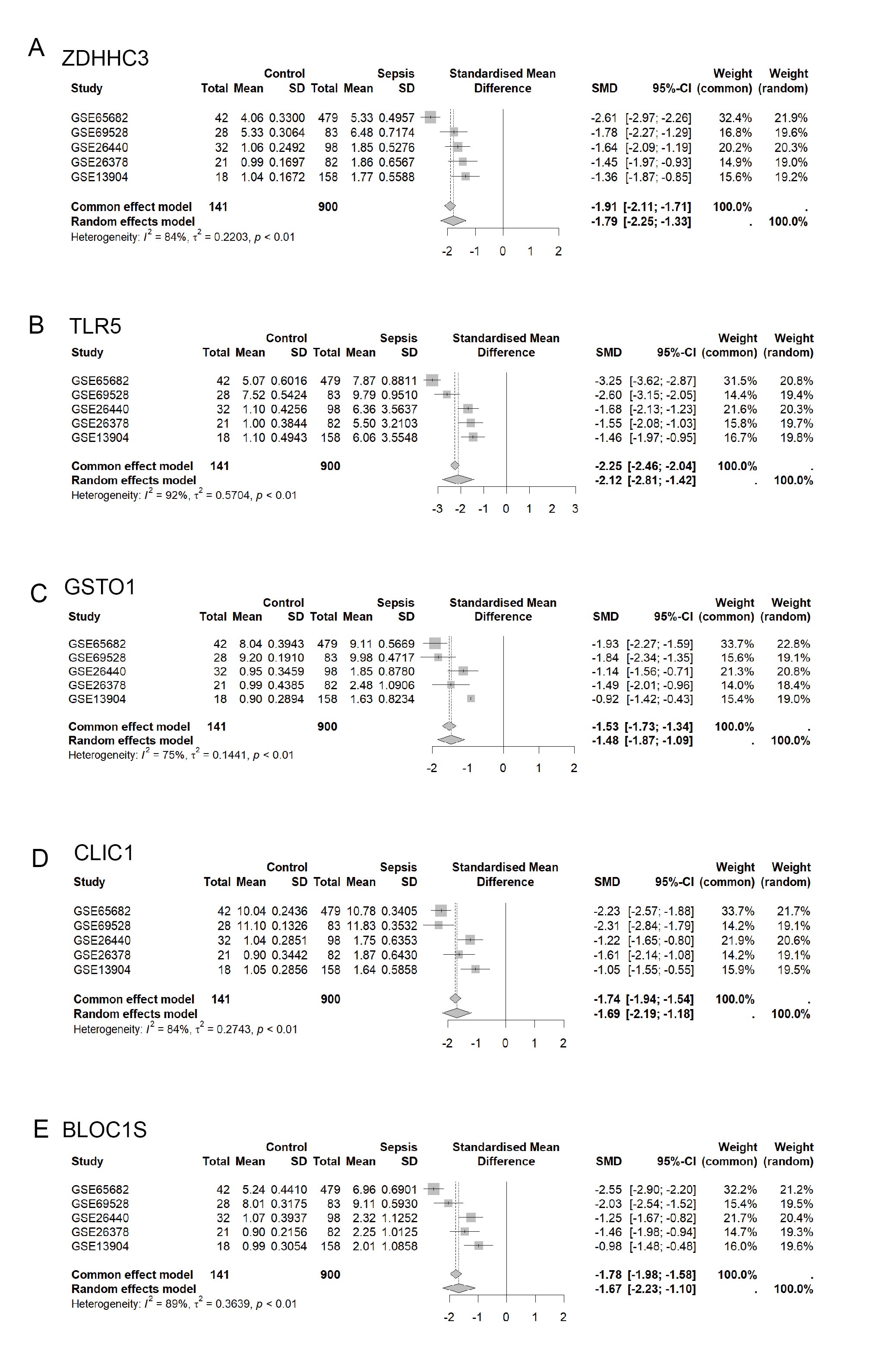{kind=link}
MR analysis of cis-eQTL of ZDHHC3 and TLR5 and septic outcomes
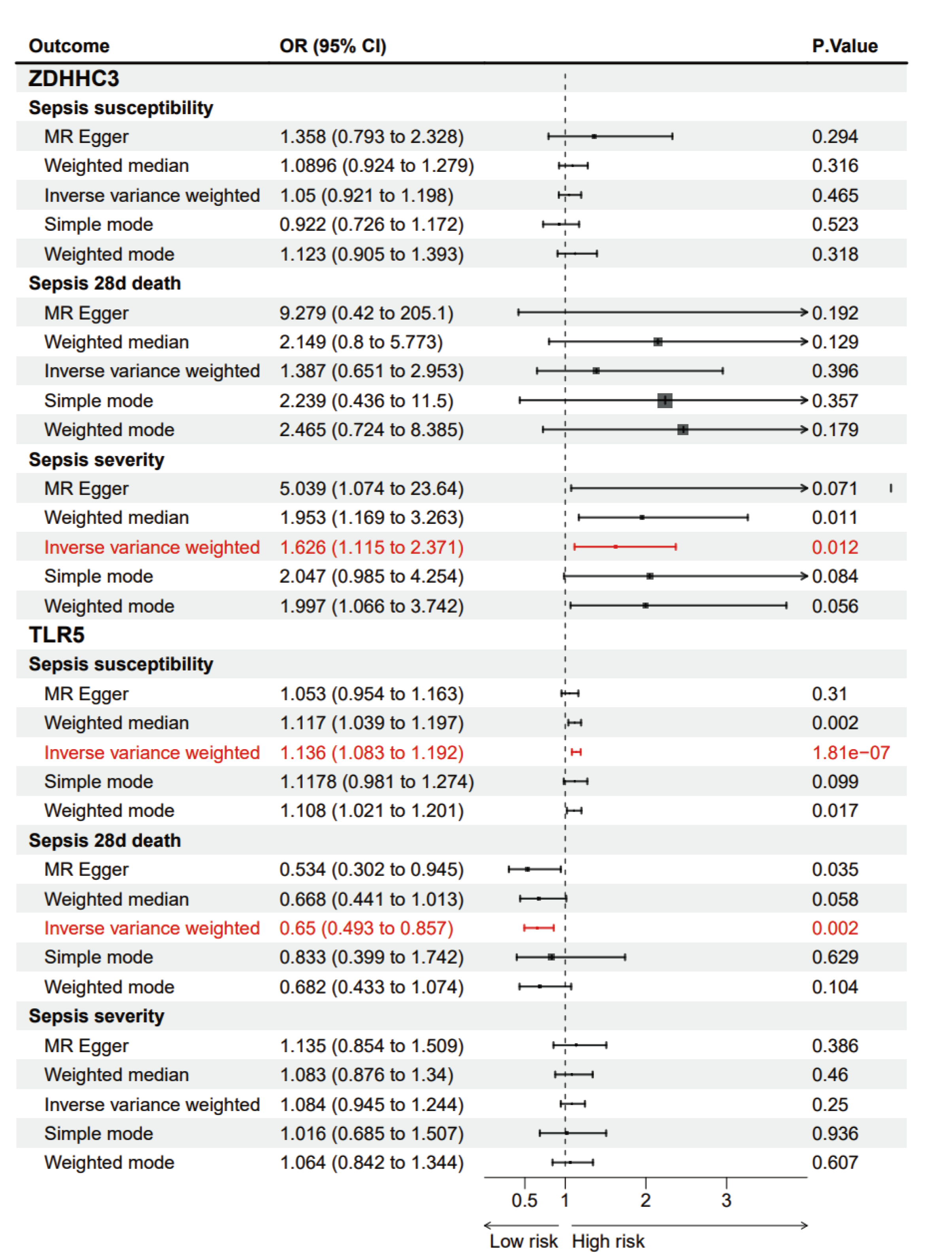{kind=link}
Sensitivity analysis
(A-C) Scatter plots of causality, detailed forest plots of each IVs, leave-one-out sensitivity analysis and funnel plots in ZDHHC3 and sepsis. (D-F) Scatter plots of causality, detailed forest plots of each IVs, leave-one-out sensitivity analysis and funnel plots in TLR5 and sepsis.
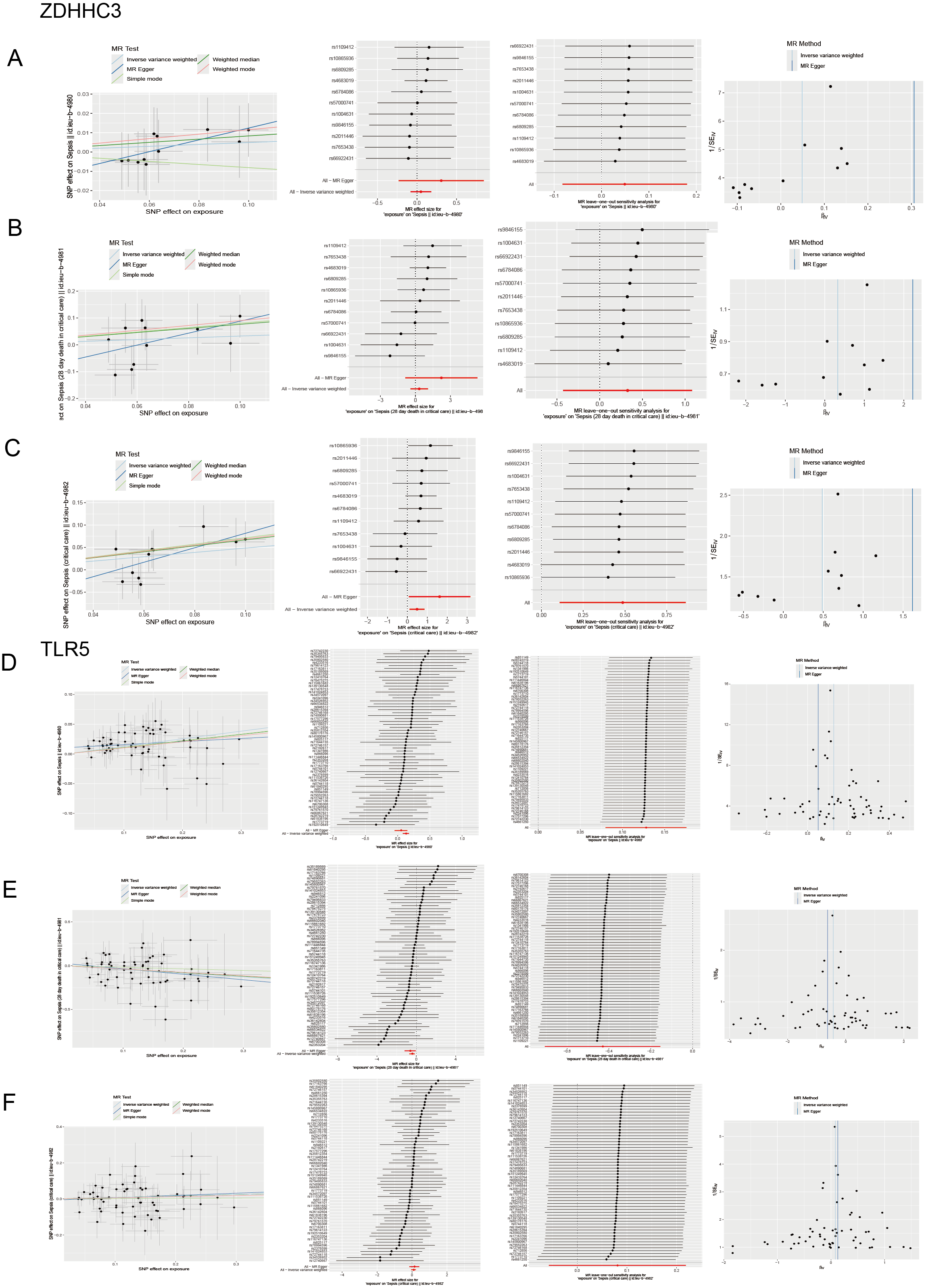{kind=link}