
Occurrence of ferredoxin:NAD+ oxidoreductase activity and its ion specificity in several Gram-positive and Gram-negative bacteria
- Published
- Accepted
- Received
- Academic Editor
- Gilles van Wezel
- Subject Areas
- Microbiology, Molecular Biology
- Keywords
- Rnf, Energy conservation, Ion pump, Fdred:NAD+ oxidoreductase
- Copyright
- © 2016 Hess et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Occurrence of ferredoxin:NAD+ oxidoreductase activity and its ion specificity in several Gram-positive and Gram-negative bacteria. PeerJ 4:e1515 https://doi.org/10.7717/peerj.1515
Abstract
A ferredoxin:NAD+ oxidoreductase was recently discovered as a redox-driven ion pump in the anaerobic, acetogenic bacterium Acetobacterium woodii. The enzyme is assumed to be encoded by the rnf genes. Since these genes are present in the genomes of many bacteria, we tested for ferredoxin:NAD+ oxidoreductase activity in cytoplasmic membranes from several different Gram-positive and Gram-negative bacteria that have annotated rnf genes. We found this activity in Clostridium tetanomorphum, Clostridium ljungdahlii, Bacteroides fragilis, and Vibrio cholerae but not in Escherichia coli and Rhodobacter capsulatus. As in A. woodii, the activity was Na+-dependent in C. tetanomorphum and B. fragilis but Na+-independent in C. ljungdahlii and V. cholerae. We deleted the rnf genes from B. fragilis and demonstrated that the mutant has greatly reduced ferredoxin:NAD+ oxidoreductase activity. This is the first genetic proof that the rnf genes indeed encode the reduced ferredoxin:NAD+ oxidoreductase activity.
Introduction
Like any other cell, microorganisms use two energy currencies to couple exergonic catabolic reactions to endergonic anabolic reactions, ATP and a transmembrane electrochemical ion gradient. Both are connected by the ATP synthase, a key element in cellular bioenergetics present in all domains of life. Under “respiratory” conditions, the ATP synthase is driven by the electrochemical ion gradient across the membrane to synthesize ATP (Boyer, Cross & Momsen, 1973). The coupling ion is a proton in most cases but some bacteria and archaea use Na+ instead of H+ as the coupling ion (Grüber et al., 2014; Müller et al., 2005). The reaction is freely reversible and under fermentative conditions ATP hydrolysis drives the generation of an electrochemical ion gradient across the membrane. Bacteria and archaea have evolved a fascinating repertoire of enzymes that generate the electrochemical ion gradient. The respiratory chain of many aerobic bacteria is the same as in mitochondria, i.e. involves complexes I, II, III, and IV (Schägger, 2002). The redox span covered by these complexes is from −320 mV (NADH/NAD+) to +800 mV (O2/H2O) and the energy released is sufficient to synthesize at least three molecules of ATP. In contrast, the facultative anaerobe Escherichia coli has a different composition and lacks a complex III (Anraku & Gennis, 1987).
Many strictly anaerobic bacteria such as, for example fermenting clostridia, acetogens or non-acetogens do not even have one of these complexes but rely on other enzyme systems to energize the membrane. In recent years, a novel type of ion-translocating redox reaction was discovered. Inverted membrane vesicles of the anaerobic acetogenic bacterium Acetobacterium woodii catalyzed electron transfer from reduced ferredoxin to NAD+ with a primary and electrogenic export of Na+ from the cytoplasm to the exterior of the cell (Biegel & Müller, 2010). Early experiments designed to verify a sodium ion dependence failed, likely due to the fact that an artificial electron donor was used in the enzymatic assay. Later, the CO dehydrogenase (CODH) was purified from A. woodii. This enzyme is able to reduce the ferredoxin purified from Clostridium pasteurianum (Hess, Schuchmann & Müller, 2013). Using C. pasteurianum ferredoxin reduced by CODH from A. woodii as electron donor, a clear dependence of ferredoxin-dependent NAD+ reduction on Na+ was observed (Hess, Schuchmann & Müller, 2013). Using the same assay, the presence of a sodium ion-dependent ferredoxin:NAD+ oxidoreductase activity was demonstrated in another acetogen, Eubacterium limosum (Jeong et al., 2015).
Moreover, using inverted membrane vesicles of A. woodii, it was demonstrated that the reaction is reversible: ATP hydrolysis drives the generation of a transmembrane electrochemical sodium ion gradient that then drives endergonic, reverse electron flow from NADH to ferredoxin (Hess, Schuchmann & Müller, 2013). The forward reaction is essential for growth of A. woodii on H2 + CO2 and fructose, but the reverse reaction is also essential, although under different growth conditions. During heterotrophic growth on many substrates such as 2,3-butanediol, lactate or ethanol, NADH is the only reductant generated. However, reduced ferredoxin is essential for the operation of the Wood-Ljungdahl pathway (Ragsdale, 2008; Schuchmann & Müller, 2014) and under these conditions, ferredoxin is reduced with NADH as reductant at the expense of the transmembrane electrochemical Na+ gradient generated by hydrolysis of ATP synthesized by substrate level phosphorylation (Hess et al., 2015; Weghoff, Bertsch & Müller, 2015). A. woodii is a prime example of an organism in which the ferredoxin:NAD+ oxidoreductase activity is essential in both directions, depending on the substrate used for growth.
The enzyme mediating the ferredoxin:NAD+ oxidoreductase activity was partially purified and amino acid sequences were derived, which allowed to identify the encoding genes. Interestingly, the encoding genes were annotated as rnf genes, based on their similiarity to the rnf genes of Rhodobacter capsulatus (Biegel, Schmidt & Müller, 2009). Deletion of the rnf genes in this phototrophic, diazotrophic bacterium led to the inability of the bacterium to fix nitrogen (Schmehl et al., 1993). The similarity of the Rnf proteins to subunits of the membrane-integral, ion translocating NADH:quinone-oxidoreductase (Biegel et al., 2011; Kumagai et al., 1997) led the authors to speculate that the Rnf proteins may constitute a membrane-integral protein complex involved in nitrogen fixation (Schmehl et al., 1993). The nitrogenase reaction requires low potential electrons that can only be generated from NADH at the expense of energy and it was speculated that the Rnf complex catalyzes this endergonic electron transfer with the energy derived from the transmembrane electrochemical ion gradient. Thus, in R. capsulatus, energy conservation is by photosynthetic electron transport phosphorylation and the Rnf complex is thought to be involved in anabolic reactions by providing low potential electrons for biosynthetic reactions (Schmehl et al., 1993).
rnf genes are widely distributed in bacteria and are also present in a few methanogenic archaea (Biegel et al., 2011). Rnf complexes thus far have not been purified from any source and thus, the final biochemical proof that they are indeed ion-translocating membrane proteins is still missing. Moreover, the anticipated reaction catalyzed by the complex, electron transfer from reduced ferredoxin to the acceptor NAD+ has only been reported for A. woodii and C. tetanomorphum (Biegel et al., 2011; Boiangiu et al., 2005). Despite the obvious lack of experimental data with a purified enzyme, as well as the experimental proof that Rnf in the methanogen Methanosarcina acetivorans uses not NAD+ but an alternative electron acceptor (Schlegel et al., 2012), the presence of rnf genes in bacteria is often taken as indication that these organisms have an ion-translocating ferredoxin:NAD+ oxidoreductase. To shed more light on the function of the Rnf complexes in different bacteria, we have isolated the cytoplasmic membranes of several organisms and assayed them for ferredoxin:NAD+ oxidoreductase activity. In addition, we determined the ion specificity of the reaction in the different organisms and will present genetic evidence that the ferredoxin:NAD+ oxidoreductase activity in B. fragilis is indeed catalyzed by the Rnf complex.
Materials and Methods
Bacterial strains and growth conditions
A. woodii DSM 1030 was grown anaerobically at 30 °C on either 20 mM fructose, 20 mM 2,3-butanediol, 50 mM ethanol, or 80 mM D,L-lactate as carbon source in 4 × 500 ml medium as described previously (Heise, Müller & Gottschalk, 1992). Clostridium ljungdahlii DSM 13528 was grown at 37 °C on 56 mM fructose as carbon source in either 2 × 500 ml (for the preparation of membranes) or 20 l (for the preparation of inverted membrane vesicles) complex medium that was prepared as described (Tanner, Miller & Yang, 1993) with slight modifications: 1000 ml medium contained: 20 g MES, 0.5 g Cysteine-HCl, 0.5 g yeast extract, 10 g fructose, 0.25 g KH2PO4, 2.5 g NH4Cl, 0.1 g CaCl2 × 2 H2O, 0.25 g KCl, 0.5 g MgSO4 × 7 H2O, 2 g NaCl, 20 mg nitrilotriacetic acid, 10 mg MnSO4 × H2O, 8 mg Fe(NH4)(SO4)2 × 6 H2O, 2 mg CoCl2, 10 mg ZnSO4 × 7 H2O, 0.2 mg CuCl2, 2 mg NiCl2 × 6 H2O, 0.2 mg Na2MoO4 × 2 H2O, 1 mg Na2SeO4 × 5 H2O, 2 mg Na2WO4, 20 μg biotin, 50 μg Ca-panthothenat, 20 μg folic acid, 50 μg liponic acid, 50 μg nicotinic acid, 100 μg pyridoxine-HCl, 50 μg p-aminobenzoic acid, 50 μg riboflavin, 50 μg thiamin-HCl, 1 μg vitamin B12, 1 mg resazurin, pH 6.5. Clostridium tetanomorphum DSM 4474 was grown at 37 °C on 222 mM glutamate in 2 × 500 ml medium that was prepared as described (Jayamani, 2008). E. coli BL21(DE3) was grown at 37 °C in 1,500 ml LB medium (Sambrook, Fritsch & Maniatis, 1989) under rigorous shaking (150 rpm). R. capsulatus SB1003 was grown at 30 °C under diazotrophic conditions in 4 × 500 ml medium, either phototrophic in N-free RCVB medium (Weaver, Wall & Gest, 1975) containing 30 mM malate as carbon source or respiratory in the dark in N-free CA medium (Saeki & Kumagai, 1998) containing 20 mM glucose and 80 mM DMSO. Bacteroides fragilis was grown in BHIS broth (Brigham & Malamy, 2005) at 37 °C in a Coy anaerobic chamber. E. coli Δrnf was grown under different conditions: under aerobic conditions in LB (Miller) medium, LBNT LB-Miller medium (including 300 mM NaCl, 50 mM Tris-HCl, pH 8), LBK medium (LB-Miller in the presence of 50 mM KCl), minimal glucose medium (M9) and in the presence of 50 mM H2O2 and 1 mM paraquat, as well as under anaerobic conditions in minimal medium (M9) containing 0.04% yeast extract with either 27 mM glycerol or 50 mM Na-formate as electron donor and different electron acceptors (40 mM nitrate, 5 mM nitrite, 70 mM DMSO, 45 mM TMAO, or 50 mM fumarate). V. cholerae was grown in LB medium (pH 7) containing 170 mM NaCl.
Preparation of washed membranes
Membranes were prepared as described (Imkamp et al., 2007) under strictly anaerobic conditions with some modifications. Cells were grown to the end of the exponential growth phase, harvested by centrifugation (11,300 × g at 4 °C; Contifuge Strato; Heraeus, Osterode, Germany) and washed twice with buffer A (50 mM Tris-HCl (pH 7.0), 20 mM MgSO4, 20% glycerol, 2 mM DTE (dithioerythritol) and 4 μM resazurin). The cells were resuspended in 15 ml buffer A and disrupted by a single passage through a French press (16,000 psi) or for B. fragilis by sonication under anaerobic conditions. Cell debris and whole cells were removed by one centrifugation step (23,700 × g, 20 min, 4 °C). The cell free extract was separated into cytoplasmic and membrane fraction by ultracentrifugation (190,000 × g, 1 h, 4 °C). The resulting sediment was washed in buffer A and membranes were again sedimented by an ultracentrifugation step (190,000 × g, 1 h, 4 °C). Membranes were resuspended in 5 ml buffer A and used immediately for the experiments. Protein concentrations were determined as described previously (Bradford, 1976).
Preparation of inverted membrane vesicles of C. ljungdahlii
Cells were grown in a 20-l-scale as described above in MES-buffered complex medium containing 56 mM fructose. 20 g (wet weight) cells were resuspended in ∼150 ml buffer A (50 mM Tris, 20 mM MgSO4, 20% glycerol, 2 mM DTE, 4 μM resazurin, pH 7.5) and digested with 400 mg lysozyme at 37 °C for 20 minutes. Afterwards, the cells were passed once through a French Press at 41 MPa. A low speed centrifugation step (4500× g, 45 minutes, 4 °C) was succeeded by ultracentrifugation at 120,000 × g and 4 °C for another 45 minutes. The vesicles were washed once in buffer A and vesicles were sedimented again via ultracentrifugation as described above. The resulting pellet was resuspended in the same buffer in a volume of 5 ml.
Measurement of Fdred:NAD+ oxidoreductase activity
Measurement of electron transfer from reduced ferredoxin to NAD+ by either membranes or inverted membrane vesicles was performed as described (Hess, Schuchmann & Müller, 2013) in anaerobic cuvettes filled with 1 ml 20 mM Tris-HCl buffer (pH 7.7) containing 2 mM DTE and 4 μM resazurin at a pressure of 0.5 × 105 Pa CO. Ferredoxin (30 μM; purified from C. pasteurianum as described (Schönheit, Wäscher & Thauer, 1978)), Acs/CODH (30 μg/ml; purified from A. woodii as described (Hess, Schuchmann & Müller, 2013)), and washed membranes or inverted membrane vesicles (150 μg/ml) were added. If indicated, the ionophores ETH2120 or TCS were added at a concentration of 10 μM. The reaction was started by addition of NAD+ (4 mM). Formation of NADH was measured at 340 nm. Enzyme activity was measured at the same temperature at which the corresponding cells were grown. Na+ dependence of the electron transfer activity was measured as described (Hess, Schuchmann & Müller, 2013).
Generation of the deletion mutant B. fragilis Δrnf
We used the two-step procedure as previously described (Brigham & Malamy, 2005) for the isolation of chromosomal deletions in broad specificity hexokinase genes of B. fragilis. Briefly, a 480 bp PCR fragment beginning 300 bp upstream of rnfB and ending 180 bp into rnfB, was ligated to a 590 bp PCR fragment containing 260 bp of the 3′ end rnfA and 330 bp of adjacent sequence, then ligated into the cloning vector pYT102 to create plasmid pRAG394 in E. coli. A tri-parental mating was performed to transfer the pRAG394 construct into the chromosome of B. fragilis ADB77. The resulting co-integrates were verified by PCR analysis and candidates were subjected to the resolution protocol as described (Brigham & Malamy, 2005). Resolvants that contained the rnf deletion were identified by PCR analysis.
Generation of the deletion mutant E. coli Δrnf
The deletion of the rnf operon from the genome of E. coli was carried out using the Red-recombinase method as reported (Datsenko & Wanner, 2000). The parent strain was E. coli TOP-10 (Invitrogen). The rnf operon was replaced with a chloramphenicol cassette, which remained in the bacterial chromosome; thus the deletion strain is resistant to chloramphenicol up to 30 μg/ml. The deletion of the rnf operon was confirmed by PCR and by verifying the loss of fluorescent bands corresponding to RnfD and RnfG in SDS-PAGE membrane preparations.
Results
For the survey of ferredoxin:NAD+ oxidoreductase activity we used membranes of the strict anaerobes A. woodii, C. tetanomorphum, C. ljungdahlii, the nanoaerophile B. fragilis, and the facultative aerobes R. capsulatus, V. cholerae and E. coli. The genetic organization of the rnf genes in these species is shown in Fig. 1.
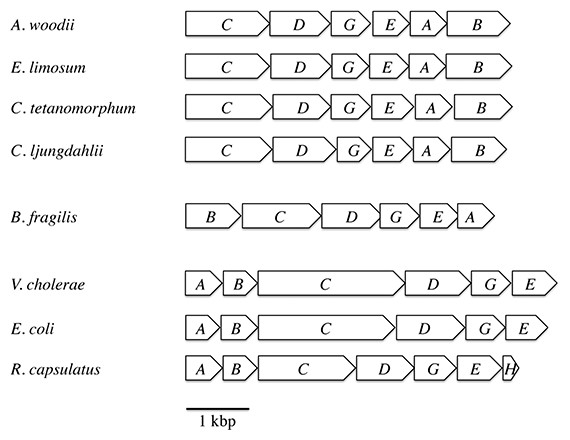
Figure 1: Arrangement of the rnf genes in different bacteria.
{kind=link}
A. woodii
As mentioned above, the acetogenic bacterium uses the Rnf complex as a membrane potential generator during growth on H2 + CO2 or fructose; and in the reverse reaction to drive the endergonic reduction of ferredoxin with electrons derived from NADH during heterotrophic growth on, for example, 2,3-butanediol, ethanol or lactate. As can be seen from Table 1, NAD+ reduction occurred at a rate of 50 ± 5 mU/mg protein and 53 ± 3 mU/mg protein, respectively, in membranes from cells grown on fructose or 2,3-butanediol, but the activity was highest in cells grown on ethanol 169 ± 8 mU/mg) or lactate (398 ± 21 mU/mg).
| Organism | Growth substrate | Fno activity without NaCl [mU/mg] | Fno activity with NaCl [mU/mg] |
|---|---|---|---|
| A. woodii | fructose | 17 ± 3a | 50 ± 5a |
| A. woodii | 2,3-butanediol | 16 ± 3b | 53 ± 3b |
| A. woodii | ethanol | 68 ± 15b | 169 ± 8b |
| A. woodii | D,L-lactate | 133 ± 19b | 398 ± 21b |
| E. limosum | fructose | 111 ± 14c | 334 ± 26c |
| C. tetanomorphum | glutamate | 421 ± 15b | 900 ± 29b |
| C. ljungdahlii | fructose | 306 ± 7b | 256 ± 7b |
| B. fragilis | glucose + brain heart infusion | 5.3 ± 0.7b | 37 ± 5b |
| B. fragilis Δrnf | glucose + brain heart infusion | 4.2 ± 0.6b | 4 ± 0.6b |
| E. coli | yeast extract | n.d. | 0b |
| R. capsulatus | glucose + DMSO | n.d. | 0b |
| R. capsulatus | malate (phototrophic) | n.d. | 0b |
| V. cholerae | LB medium | 8.2 ± 0.8b | 7.1 ± 0.7b |
Notes:
Fno, ferredoxin; NAD+, oxidoreductase activity; n.d., not determined. Each value is the mean from 3 replicates.
C. ljungdahlii
C. ljungdahlii can grow autotrophically on H2 + CO2 or CO and produces acetate and ethanol. Inhibitor studies that had been done with whole cells were consistent with the presence of an enzyme that generates a primary electrochemical proton potential across the cytoplasmic membrane (Tremblay et al., 2012). Deletion of the rnf genes had resulted in a loss of the proton motive force which is consistent with the hypothesis of the presence of a proton-translocating Rnf complex in C. ljungdahlii. Indeed, membranes of C. ljungdahlii catalyzed electron transfer from reduced ferredoxin to NAD+ with an activity of 256 ± 7 mU/mg (Table 1). This activity was not dependent on Na+ (measured in a range of 68 μM to 10 mM NaCl) (Fig. 2A). Electron transfer from reduced ferredoxin to NAD+ in inverted membrane vesicles was stimulated by the protonophore TCS but not by the sodium ionophore ETH2120 (Fig. 2B). These data are consistent with the hypothesis that the Rnf complex of C. ljungdahlii uses H+ as coupling ion, not Na+. F1FO (Rahlfs & Müller, 1997; Müller, 2003) and A1AO (Müller, Rupert & Lemker, 1999) ATP synthases have a characteristic Na+ binding motif in their c subunits (Q/E in helix one, E/D and S/T in helix two). The absence of a Na+-binding motif in subunit c of the ATP synthase in C. ljungdahlii is in line with the hypothesis of a H+-Rnf.
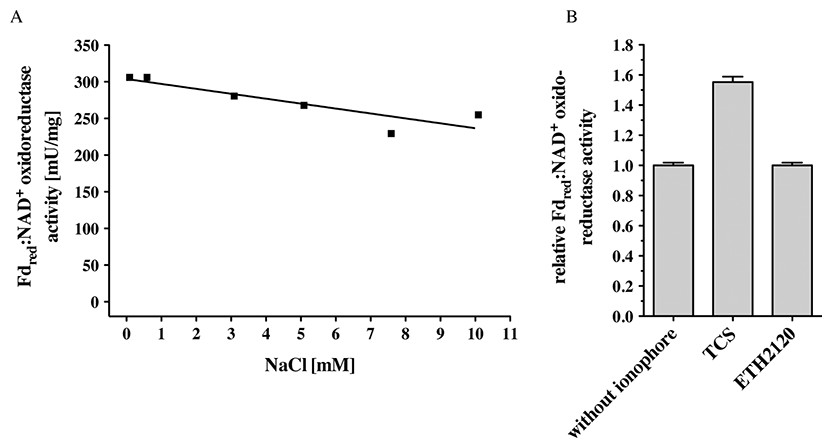
Figure 2: Fdred:NAD+ oxidoreductase activity of membranes of C. ljungdahlii as a function of Na+ concentration (A) and of inverted membrane vesicles in the presence of different ionophores (B).
Fno activity was measured in anoxic cuvettes filled with 1 ml 20 mM Tris (sodium free)-HCl buffer (pH 7.7) containing 2 mM DTE and 2 μM resazurin at a pressure of 0.5 × 105 Pa CO. NaCl was added to the concentration indicated. Ferredoxin (30 μM), Acs/CODH (30 μg/ml), and washed membranes or inverted membrane vesicles (150 μg/ml) were added. If indicated, the ionophores ETH2120 or TCS were added at a concentration of 10 μM. The reaction was started by addition of NAD+ (4 mM). Formation of NADH was measured at 340 nm.{kind=link}
C. tetanomorphum
This obligate anaerobic, glutamate fermenting bacterium was quoted in review articles to have an Rnf complex that could be partially purified (Boiangiu et al., 2005; Buckel & Thauer, 2013). The ion specificity was not discussed but speculated to be Na+. However, these data are not published. Membranes of C. tetanomorphum grown on glutamate catalyzed electron flow from reduced ferredoxin to NAD+ with a rate of 900 ± 29 mU/mg (Table 1), 18-fold higher than the rate observed in fructose-grown cells of A. woodii. Without additional Na+ the activity was only 431 ± 16 mU/mg, which is consistent with the hypothesis that the enzyme uses Na+ as coupling ion.
E. coli
The rnf genes in E. coli are termed rsx (Koo et al., 2003) and have been shown to be upregulated during aerobic growth (Partridge et al., 2006). However, membranes from aerobically grown cells prepared under anaerobic conditions had no ferredoxin:NAD+-oxidoreductase activity. Furthermore, a deletion of the rnf operon from E. coli in the strain TOP10 showed no growth phenotype, neither under aerobic nor under anaerobic conditions.
R. capsulatus
As mentioned above, the rnf genes were first described in R. capsulatus and hypothesized to be involved in nitrogen fixation by providing low potential electron for nitrogenase (Schmehl et al., 1993). Membranes of cells grown diazotrophically had no ferredoxin:NAD+-oxidoreductase, neither if the cells were grown in the light nor under respiratory conditions with glucose or DMSO.
B. fragilis
The nanoaerophilic bacterium B. fragilis not only has rnf genes but also nqr and nuo (complex I) genes. Cytoplasmic membranes prepared from cells grown on complex medium catalyzed electron transfer from reduced ferredoxin to NAD+ with a rate of 37 ± 5 U/mg (Table 1). In the absence of NaCl, the activity was only 5.3 ± 0.7 mU/mg (Table 1), arguing for a Na+-dependence of the reaction. Since a genetic system is available for this organism, we deleted the rnf genes to establish the gene-polypetide correlation of the observed activity. Indeed, the ferredoxin:NAD+ oxidoreductase activity was reduced to 11% in the Δrnf mutant in both the presence and absence of NaCl (Table 1), which is evidence that the majority of the ferredoxin:NAD+ oxidoreductase measured is encoded by the rnf genes.
V. cholerae
The presence of rnf genes in V. cholerae was long known, however, a function of Rnf in this bacterium has not yet been described. Cytoplasmic membranes of V. cholerae catalyzed electron transfer from reduced ferredoxin to NAD+ with a rate of 7.1 ± 0.7 mU/mg (Table 1). As in C. ljungdahlii, this activity was independent of the Na+ concentration, arguing for a H+-dependent enzyme.
Discussion
The assay described before to detect ferredoxin:NAD+ oxidoreductase activity in A. woodii (Hess, Schuchmann & Müller, 2013) was apparently suitable to detect the same activity in C. ljungdahlii, C. tetanomorphum, B. fragilis, and V. cholerae. This is actually all the more surprising since the assay requires that the ferredoxin:NAD+ oxidoreductase accepts electrons from C. pasteurianum ferredoxin. Not only must the redox potential be suitable but also the protein-protein interaction of the ferredoxin and its cognate receptor, most likely RnfB (Suharti et al., 2014) has to allow electron flow from the donor protein to the acceptor protein. The lack of activity in membranes of E. coli or R. capsulatus may be due to the differences in ferredoxin or simply reflect the fact that ferredoxin is not the electron carrier used by these enzyme complexes. Bacteria such as A. woodii encode several different ferredoxins but the one used by the ferredoxin:NAD+ oxidoreductase is not known. Therefore, phylogenetic analysis cannot be done. Thus far, the electron acceptor used by the Rnf complexes of E. coli and R. capsulatus remain to be identified. Moreover, the Δrnf strain of E. coli has no phenotype and thus, the physiological role in E. coli also remains to be elucidated. Koo et al. (2003) has suggested that Rnf is the electron donor to the Fe-S protein SoxR that is part of a ROS sensor in the cell (Hidalgo, Ding & Demple, 1997). The role of Rnf would be to maintain SoxR in a reduced, and thus inactive state. In the presence of oxygen radicals, oxidized SoxR signals the cell to activate a response to ROS thus in the rnf deletion mutant, SoxR is expected to always be in the oxidized state and the cellular systems that protect the cell against ROS would always be activated. This would make it difficult to define phenotypic differences between the rnf deletion mutant and wild type E. coli. In our hands the growth curve for the deletion mutant was essentially the same as for wild type in both rich and minimal media, and inclusion of ROS producing substances such as paraquat and hydrogen peroxide did not reveal any clear differences. Preliminary expression data for V. cholerae suggest that Rnf is expressed at the same levels in several growth and infection conditions, suggesting that changes in the environment have little effect on the expression of rnf. However, Rnf seems to be essential for V. cholerae since we were not able to generate a Δrnf deletion mutant.
Of interest is the finding that the rnf deletion in B. fragilis did not alter the anaerobic growth properties of this strain on BHIS medium (which contains glucose) when compared to the wild type parental strain. There are other annotated oxidoreductases in the B. fragilis genome coded for by the nuo and nqr operons whose functions may be redundant with rnf; this might also explain why 11% of the ferredoxin:NAD+ oxidoreductase activity could still be measured in the rnf deletion strain.
The data presented here show that the ferredoxin:NAD+ oxidoreductase activity in A. woodii is regulated, since growth on different substrates resulted in different activities. There was no correlation between activity and direction of electron flow in context of the metabolism. However, there was a correlation between activity and the number of cycles the complex has to pass for the phosphorylation of 1 ATP in the metabolism: When converting lactate, the Rnf complex has to catalyze 2.5 cycles for every ATP that is gained in this pathway (Weghoff, Bertsch & Müller, 2015). When converting ethanol, the phosphorylation of 1 ADP requires the Rnf complex to catalyze 1.3 cycles (Bertsch et al., 2015). The conversion of 2,3-butanediol only needs 0.85 cycles for every ATP (Hess et al., 2015) and degradation of fructose is predicted to require 0.11 Rnf cycles per ATP. The correlation of these values is in good agreement with the specific Rnf activities we observed for each of these substrates (Table 1). Thus, the more Rnf activity is necessary for the metabolism, the higher is the experimentally determined activity.
The highest ferredoxin:NAD+ oxidoreductase activity was found in C. tetanomorphum. Again, this would argue for a high electron transfer rate through Rnf during glutamate fermentation. The activity was stimulated by Na+ indicating that the Rnf complex uses Na+ as coupling ion. This is also consistent with the hypothesis of a Na+/glutamate symporter in C. tetanomorphum (Buckel & Thauer, 2013) as well as a V-type ATP synthase (encoded in the genome) with the c subunit having a Na+ binding motif in the second hairpin. In contrast, the activity was not Na+-dependent in the acetogen C. ljungdahlii. The observed stimulation of NAD+ reduction in inverted membrane vesicles by a protonophore clearly resembles the phenomenon of respiratory control, showing a strict coupling of electron transfer to the translocation of an ion across the membrane. The electrochemical potential established by the ion and charge translocation sets up a thermodynamic backup pressure and reduces the rate of electron flow (as well as the coupled ion flow). Dissipation of the ion gradient will release the thermodynamic backup pressure and stimulate electron flow. Since this was observed only with a protonophore but not a sodium ionophore it can be concluded that the coupling ion is a proton, not a sodium ion. As discussed before (Biegel et al., 2011), there is no reason to believe that the Rnf complex can only translocate Na+ as it does in A. woodii. Actually, other membrane transporters are known that can translocate either Na+ or H+, depending on the species. In the case of the F1FO ATP synthase, only the exchange of two out of roughly 4,000 residues will change the ion specificity of the enzyme from H+ to Na+ (Meier et al., 2009).
The final proof that the Rnf complex is a redox-driven ion (Na+/H+ pump) still has to await purification of the complex and its reconstitution into liposomes. An equally important proof is presented here for the first time in bacteria: deletion of the rnf genes of B. fragilis reduced the ferredoxin:NAD+ oxidoreductase activity by 89%. Although the physiological role of the Rnf complex and the origin of the remaining 11% activity remains to be identified, this is clear evidence that the rnf genes encode the ferredoxin:NAD+ oxidoreductase activity and in line with the observation that a Δrnf mutant of the archaeon Methanosarcina acetivorans is impaired in Na+ transport coupled to electron flow from reduced ferredoxin to heterodisulfide (Schlegel et al., 2012).