
Screening of high-risk deleterious missense variations in the CYP1B1 gene implicated in the pathogenesis of primary congenital glaucoma: A comprehensive in silico approach
- Published
- Accepted
- Received
- Academic Editor
- Luisa Azevedo
- Subject Areas
- Bioinformatics, Computational Biology, Genomics, Molecular Biology, Ophthalmology
- Keywords
- Primary congenital glaucoma, CYP1B1, Missense variations, In silico, Modeling, Molecular dynamic simulation
- Copyright
- © 2022 Shahid et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2022. Screening of high-risk deleterious missense variations in the CYP1B1 gene implicated in the pathogenesis of primary congenital glaucoma: A comprehensive in silico approach. PeerJ 10:e14132 https://doi.org/10.7717/peerj.14132
Abstract
Background
Primary congenital glaucoma (PCG) is the most common subtype of glaucoma caused by defects in the cytochrome P450 1B1 (CYP1B1) gene. It is developing among infants in more than 80% of cases who exhibit impairments in the anterior chamber angle and the trabecular meshwork. Thus, a comprehensive in silico approach was performed to evaluate the effect of high-risk deleterious missense variations in the CYP1B1 gene.
Material and methods
All the information for CYP1B1 missense variants was retrieved from the dbSNP database. Seven different tools, namely: SIFT, PolyPhen-2, PROVEAN, SNAP2, PANTHER, PhD-SNP, and Predict-SNP, were used for functional annotation, and two packages, which were I-Mutant 2.0 and MUpro, were used to predict the effect of the variants on protein stability. A phylogenetic conservation analysis using deleterious variants was performed by the ConSurf server. The 3D structures of the wild-type and mutants were generated using the I-TASSER tool, and a 50 ns molecular dynamic simulation (MDS) was executed using the GROMACS webserver to determine the stability of mutants compared to the native protein. Co-expression, protein-protein interaction (PPI), gene ontology (GO), and pathway analyses were additionally performed for the CYP1B1 in-depth study.
Results
All the retrieved data from the dbSNP database was subjected to functional, structural, and phylogenetic analysis. From the conducted analyses, a total of 19 high-risk variants (P52L, G61E, G90R, P118L, E173K, D291G, Y349D, G365W, G365R, R368H, R368C, D374N, N423Y, D430E, P442A, R444Q, F445L, R469W, and C470Y) were screened out that were considered to be deleterious to the CYP1B1 gene. The phylogenetic analysis revealed that the majority of the variants occurred in highly conserved regions. The MD simulation analysis exhibited that all mutants’ average root mean square deviation (RMSD) values were higher compared to the wild-type protein, which could potentially cause CYP1B1 protein dysfunction, leading to the severity of the disease. Moreover, it has been discovered that CYP1A1, VCAN, HSD17B1, HSD17B2, and AKR1C3 are highly co-expressed and interact with CYP1B1. Besides, the CYP1B1 protein is primarily involved in the metabolism of xenobiotics, chemical carcinogenesis, the retinal metabolic process, and steroid hormone biosynthesis pathways, demonstrating its multifaceted and important roles.
Discussion
This is the first comprehensive study that adds essential information to the ongoing efforts to understand the crucial role of genetic signatures in the development of PCG and will be useful for more targeted gene-disease association studies.
Introduction
Glaucoma is a heterogeneous group of complex diseases that gradually lead to permanent loss of vision. It is estimated that 79.6 million people worldwide were affected in 2020, and the number is predicted to rise to 111.8 million by 2040 (Quigley & Broman, 2006; Tham et al., 2014). The modern classification categorizes glaucoma into three groups, which are: primary congenital glaucoma (PCG); primary open-angle glaucoma (POAG); and primary angle closure glaucoma (PACG). The most common subtype of glaucoma is PCG, and it has been identified as developing among infants in more than 80% of cases. This disorder develops due to impairments in the anterior chamber angle and the trabecular meshwork. Clinically, the affected patients manifest corneal edema, photophobia, epiphora (watery eye), and buphthalmolos (enlargement of the globe), which result from elevated intraocular pressure (IOP) (Vasiliou & Gonzalez, 2008). The increased of IOP in untreated individuals with PCG is a major risk factor that leads to rapid axonal loss and permanent loss of vision (Vasiliou & Gonzalez, 2008). The incidence rate of PCG are varies depending on ethnic and geographical groups, where 1:1250 has been reported in the Slovak Roman population; 1:10000 in Western countries and 1:2500 in the Middle East (Badawi et al., 2019; Gencik, Gencikova & Ferak, 1982).
PCG is an autosomal recessive disorder that has a variable penetrance rate. It more frequently affects males at around 65% compared to females at 35%, demonstrating the sporadic transmission (in 90% of cases), but pseudodominant transmission has also been shown in some families (Bejjani et al., 2000). Through genetic linkage analysis, four loci of PCG have been discovered, which were GLC3A, GLC3B, GLC3C, and GLC3D, on chromosomes 2p22-p21, 1p36.2-p36.1, 14q24.3, and 14q24, respectively (Akarsu et al., 1996; Firasat et al., 2008; Sarfarazi et al., 1995; Stoilov, MJIo & v, 2002). In addition, two genes, CYP1B1 (coding for cytochrome P450, family 1, subfamily B, polypeptide 1) and, LTBP2 (coding for latent transforming growth factor β-binding protein 2) were recently discovered. CYP1B1 was found on GLC3A loci, while LTBP2 was found on GLC3D loci (Ali et al., 2009; Stoilov, Akarsu & Sarfarazi, 1997). Molecular screening revealed that CYP1B1 is a more mutated gene compared to LTBP2, and hundreds of variations have been reported in this gene that are associated with PCG phenotypes (Rashid et al., 2019).
CYP1B1 belongs to the CYP450 superfamily and genomic DNA length of 8.5 kb. This gene is expressed in the eye, kidney, breast, prostate, and brain tissues (Muskhelishvili et al., 2001). It is also known as the first gene predominantly associated with primary developmental defects (Muskhelishvili et al., 2001). Functionally, it is a member of the aryl hydrocarbon receptor (AHR) battery that is incorporated into both endogenous and exogenous substrate metabolism (Tang et al., 1996). It is exclusively involved in the metabolism of carcinogens, steroids, arachidonate, retinol, retinal, and melatonin that may trigger signal transduction pathways to control the growth and differentiation of tissues. Through these corresponding mechanisms, CYP1B1 probably regulates the early ocular differentiation processes. Recent studies found that CYP1B1 is also expressed constitutively in vascular cells, which play an important role during postnatal retinal vascular development (Tang et al., 2009). Apart from that, several studies conducted on humans and mice have elucidated that CYP1B1 plays a pivotal role in the proper development of trabecular meshwork and other ocular tissues. Besides PCG, CYP1B1 has also been linked with other diseases, including various types of cancer and cardiovascular diseases (Falero-Perez et al., 2018).
In the development and progression of a disease, several environmental and genetic factors are involved. Previous research found that single nucleotide polymorphisms (SNPs) are the most common genetic determinants, occurring in one out of every 3000 base pairs in the human genome and serving as potential biomarkers for a variety of life-threatening diseases (Earl & Greenhalf, 2009). Among all SNPs, the presence of non-synonymous SNPs (nsSNPs), also called missense variations, is considered to have profound phenotypic effects upon a variation in protein amino acid sequence. It has been seen that these missense variations have substantial potential to alter the structure, stability, biological functions, and interactions of the protein (Bhattacharya & Ray, 2021). Therefore, numerous in silico studies have been carried out previously to explore the deleterious missense variation in many genes associated with various diseases’ pathogenesis (Agrahari et al., 2019; Bhattacharya & Ray, 2021; Shahid et al., 2022; Sunkar & Neeharika, 2020). The identification of deleterious missense variations in the CYP1B1 gene is absolutely essential for more detailed comprehension to identify novel biomarkers in the prognosis and management of PCG. Therefore, this study aimed to investigate the structural and functional effects of the high-risk deleterious missense variants of CYP1B1 using various computational tools.
Material and Methods
Data retrieval
The list of missense variations in the CYP1B1 gene of Homo sapiens was retrieved from the dbSNP database of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/projects/SNP/). The encoded protein sequence with reference ID: NP_000095.2 was also downloaded from the same database.
Functional annotation of variants
The experiment is carried out computationally and is called the “in silico method.” These methods use databases, network analysis, and machine learning approaches that are faster, more critical, and more cost-effective than in vitro and in vivo studies (Azfaralariff et al., 2022). Therefore, in the present study, to identify the high-risk deleterious variations, the functional annotation of missense variations in the CYP1B1 gene was carried out by employing multiple bioinformatics tools. Seven different tools, which were SIFT, PolyPhen-2, PROVEAN, SNAP2, PANTHER, PhD-SNP, and Predict-SNP, were used in this screening assay. The first screening tool employed in this assay to investigate the deleterious variants from the data retrieved from the dbSNP NCBI database was called SIFT (sorting intolerant from tolerant) (https://sift.bii.a-star.edu.sg/). It predicts the functional importance of the altered amino acid substitution using evolutionary conservation via sequence homology alignment and the physiochemical characteristics of a protein. The SIFT algorithm predicts substitutions with a probability score ≥ 0.05 as tolerated substitutions, while those with a probability score <0.05 are considered deleterious (Ng & Henikoff, 2003; Shahid et al., 2020). Polyphen 2.0 (polymorphism phenotyping v2) (http://genetics.bwh.harvard.edu/pph2/) was utilized, acknowledging its structural and functional impact prediction of a single amino acid alteration in a protein. This tool evaluates an amino acid substitution using comparative homology protein models and generates a position specific independent count (PSIC) score. It annotates a substitution as probably damaging, possibly damaging, or benign if the probability score is >0.85, >0.15, and <0.15, respectively (Adzhubei et al., 2010). Further, the PROVEAN (Protein Variation Effect Analyzer) (http://provean.jcvi.org/index.php) server was accessed in this functional annotation assay. This server predicts the functional impact of an amino acid alteration in a protein and categorizes them as neutral or deleterious, if the generated score is greater than −2.5 or less than −2.5, respectively (Choi et al., 2012). The next screening was performed by the PANTHER (Protein analysis through evolutionary relationship-position-specific evolutionary preservation) (http://www.pantherdb.org/) server. This tool calculates the score of an amino acid variant based on evolutionary preservation (time in millions of years) using the HMM algorithm. It annotates an amino acid alteration as benign: time <200 my, possibly damaging: 450 my to 200 my, or probably damaging: 450 my, respectively (Tang & Thomas, 2016). SNAP2 (Screening of non-acceptable Polymorphism) (https://rostlab.org/services/snap2web/) tool was employed to determine the pathogenicity of an amino acid alteration in a protein. It is an ensemble of neural network models that predicts both the clinical and molecular effects of a missense variant in the form of strongly effective (score: +100) or strongly neutral (score: −100) (Bromberg & Rost, 2007). Moreover, the PhD-SNP (Predictor of human deleterious single nucleotide polymorphism) tool, accessed from https://snps.biofold.org/phd-snp/phd-snp.html, was also utilized in this functional assay. It is a Support Vector Machine (SVM) tool that predicts the pathogenicity of an amino acid substitution in a protein based on evolutionary information and a hybrid predictive model. This tool classifies the missense variants as neutral or pathogenic and provides a reliability index (RI) ranging from 0-10, with 10 being the highest reliability (Capriotti, Calabrese & Casadio, 2006). Additionally, the Predict-SNP (https://loschmidt.chemi.muni.cz/predictsnp/) tool was also approached to predict the disease related variations of the CYP1B1 gene. It uses consensus-based methods to generate its output (Bendl et al., 2014). The allelic frequency of the variants was determined using the gnomAD (Genome Aggregation Database) version 2.1.1 (https://gnomad.broadinstitute.org/). The gnomAD is the most widely used database that harbors the largest collection of sequencing data for population variations (Gudmundsson et al., 2022).
Effect of variations on CYP1B1 protein stability
To evaluate the stability of the protein upon any amino acid alteration, two machine learning tools, namely, I-Mutant 2.0 and MUpro, were utilized. I-Mutant 2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html) predicts protein stability using an SVM-based algorithm and calculates the amino acid change in the form of Gibbs free energy (DDG) at a given pH and temperature. It categorizes the protein stability as increased or decreased and provides a reliability index (RI) ranging from 0 to 10, with 10 being the highest reliability (Capriotti, Fariselli & Casadio, 2005). The input conditions were set to pH 7 and temperature 37 °C respectively. Additionally, the MUpro server (http://mupro.proteomics.ics.uci.edu/) was approached to validate the I-Mutant 2.0 outcomes. This server ensembles both SVM and neural network algorithms to predict the effect of amino acid alteration in a protein and classifies the protein stability as increased or decreased if the confidence scores are <0 or >0, respectively (Cheng et al., 2006).
Conservancy analysis of high-risk variations and secondary structure of CY1B1
The evolutionary conservation of the high risk deleterious amino acid variations was evaluated by utilizing the ConSurf server (https://consurf.tau.ac.il/). This server works on the empirical Bayesian method and multiple sequence alignment approach to estimate the phylogenetic conservation score, which ranges from 1–9. Residues of conservation scores of 7–9 were considered highly conserved, 4–6 as intermediate, and 1–3 were represented as variable and less conserved. In addition, this tool also predicted the characteristics of residues based on their positions in the protein’s structure: either buried (b), exposed (e), highly conserved and exposed (functional, f), or highly conserved and buried (s), which further embellished the structural and functional significance of amino acid residues in a protein (Ashkenazy et al., 2010; Shahid et al., 2022). For the secondary structure prediction of CYP1B1 protein, the SOPMA (Self-optimized Prediction Method with Alignment) bioinformatic tool was utilized by using its default query parameters (window width, 17; similarity threshold, 8; and number of states, 4). This server ensembles the self-optimized prediction method and a neural network method (PHD) to generate the secondary structure of a protein (Geourjon & Deleage, 1995).
Structural examination of native and mutant types of CYP1B1
The three-dimensional (3D) structure of CYP1B1 protein was downloaded from the Protein Data Bank (PDB) (https://www.rcsb.org/). To generate the complete structure of the CYP1B1 protein, the I-TASSER (Iterative Threading Assembly Refinement) (https://zhanggroup.org/I-TASSER/) most advanced biological server was approached (Zhang, 2008). This is a tool that constructs the 3D structures of proteins through hierarchical approaches by first searching the templates from the PDB database and then constructing the models through iterative template fragment assembly simulation. Moreover, 19 mutant structures of CYP1B1 protein were also generated through the I-TASSER tool by incorporating the amino acid alteration of each high-risk deleterious variation into the wild-type FASTA protein sequence. The quality (degree of appropriateness) of all the native and mutant structures was verified through the ERRAT and VERIFY3D, which are in-built tools of the SAVES meta-server version 6.0 (https://saves.mbi.ucla.edu/). Subsequently, the resultant structures were further subjected to the TM-Align server (https://zhanglab.dcmb.med.umich.edu/TM-align/) which calculates the RMSD (Root Mean Square Deviation) to analyze the deviation of mutant structures from the wild-type (Zhang & Skolnick, 2005). Graphical inspection and superimposition of wild-type and mutant structures were performed by UCSF Chimera software version 1.14 (Pettersen et al., 2004).
Molecular dynamics simulation (MDS) analysis
The molecular dynamic simulation (MDS) for the native and mutant CYP1B1 protein structures was carried out using GROMACS simulation software accessed from https://simlab.uams.edu (Bekker et al., 1993). For the MDS run, the wild-type and mutant protein structures were prepared using the GROMOS96 43a1 force field solvated by the Single Point Charge (SPC) water model and fixed in a periodic cubic solvated box. Other requisite parameters in molecular dynamic simulation, such as neutralization by adding the salt (NaCl) concentration (0.15 M), energy minimization for 5,000 steps of the prepared system using the steepest descent method, equilibration type consisting of NVT/NPT at 300 K and 1 bar of pressure using the leap-frog method. The final MD simulation step was processed for 50 ns to analyze the deviation of mutant structures from the wild-type protein structure.
Investigation of CYP1B1-interacting candidates
The co-expression analysis of the CYP1B1 gene was evaluated through the GeneMANIA server (https://genemania.org/). It is a publically available bioinformatics tool that harbors a large database for gene functional analysis, including co-expression, pathway interaction, physical interaction, co-localization, genetic interaction, and shared protein domains (Shahid et al., 2021; Warde-Farley et al., 2010). The submitted query was run by setting Homo sapiens as the species organism and the maximum number of resultant genes and attributes as 10. On top of that, the protein-protein interaction (PPI) of CYP1B1 was also determined by utilizing the STRING protein database (https://string-db.org/). The STRING database query terms were cut-off at 0.7 and the maximum additional interaction was 10 (Azfaralariff et al., 2022; Szklarczyk et al., 2021). Cytoscape software version 3.8.2 (https://cytoscape.org/) was employed to construct the highly co-expressed genes and PPI networks in the present study (Shannon et al., 2003).
Gene ontology and pathway enrichment analysis of CYP1B1
The functional annotation of three gene ontology (GO) terms, which were molecular functions (MF), cellular components (CC), and biological processes (BP), and the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway of CYP1B1 were analysed with Enrichr database (https://maayanlab.cloud/Enrichr/), a publically available online tool for gene expression profiling, transcriptional factor analysis, PPI networks, pathway analysis, toxicogenomics and pharmacogenomics studies (Chen et al., 2013). The P-value <0.05 was set as a cut-off criterion for the submitted query. Cytoscape software was utilized to construct all the networks.
Results
Data retrieval and selection
The dbSNP is a unique database that holds all the necessary information, including the variation data of many of the human genes. This database with CYP1B1 reported a total of 4788 SNPs, of which 610 were missense, 276 were synonymous, 1359 were in the intronic region, and the rest belonged to other categories (Accessed: December 21, 2021). In this study, the missense variations were selected in order to investigate their damaging effects and how they influence the phenotypic characteristics of the respective encoded protein. These missense variations are responsible for various complex diseases by affecting the protein’s functional activities and structural stability or folding (Khalid & Almaghrabi, 2020; Shahid et al., 2022). A list of all 610 missense variants is presented in Table S1.
Functional annotation of missense variants
The missense variations of a gene can produce amino acid allelic variants that may influence the functions of that particular gene product by disrupting the structural conformation. Hence, missense variants of the CYP1B1 gene were chosen for further investigation. Variations of 610 CYP1B1 missense retrieved from the dbSNP NCBI database were first subjected to the SIFT server, which predicted 92 variants to be either deleterious or tolerated (Table 1; Table S1 ). These 92 variants were further evaluated by six other computational tools, namely PolyPhen-2, PhD-SNP, SNAP2, PANTHER, PROVEAN, and Predict-SNP, which used different algorithms to scrutinize a variant. The predictions and categories of these tools are provided in Table 1. From conducted analysis, a total of 19 missense variants (P52L, G61E, G90R, P118L, E173K, D291G, Y349D, G365W, G365R, R368H, R368C, D374N, N423Y, D430E, P442A, R444Q, F445L, R469W, and C470Y) were found to be common in all the functional analysis tools by manual concordance, and all were deleterious, probable damaging, and effect variants. Therefore, these 19 variants were considered the high-risk deleterious variants of the CYP1B1 gene and are promising candidates for further research. The detailed results of 19 high-risk variants are presented in Table 2 and their allele frequencies information are in Table 3.
| Tool | Prediction | Software outputs |
|---|---|---|
| SIFT | Tolerated | 38 |
| Deleterious | 54 | |
| Total | 92 | |
| PolyPhen-2 | Benign | 30 |
| Possibly damaging | 15 | |
| Probably damaging | 47 | |
| Total | 92 | |
| PROVEAN | Neutral | 46 |
| Deleterious | 46 | |
| Total | 92 | |
| PANTHER | Benign | 0 |
| Possibly damaging | 50 | |
| Probably damaging | 42 | |
| Total | 92 | |
| SNAP2 | Neutral | 44 |
| Effective | 48 | |
| Total | 92 | |
| PhD-SNP | Neutral | 52 |
| Disease | 40 | |
| Total | 92 | |
| Predict SNP | Neutral | 45 |
| Deleterious | 47 | |
| Total | 92 |
| rs IDs | Variant | SIFT | PolyPhen 2 | PROVEAN | PANTHER | SNAP2 | Phd SNP | Predict SNP | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Score | Prediction | Score | Prediction | Score | Prediction | Score | Prediction | Score | Prediction | Prediction | Score | Prediction | ||
| rs201824781 | P52L | 0 | Deleterious | 1 | Probably Damaging | −6.33 | Deleterious | 0.85 | Probably Damaging | 58 | effect | Disease | 0.86908 | Deleterious |
| rs28936700 | G61E | 0 | Deleterious | 1 | Probably Damaging | −6.60 | Deleterious | 0.85 | Probably Damaging | 90 | effect | Disease | 0.86908 | Deleterious |
| rs112014184 | G90R | 0 | Deleterious | 1 | Probably Damaging | −6.94 | Deleterious | 0.85 | Probably Damaging | 91 | effect | Disease | 0.86908 | Deleterious |
| rs73625096 | P118L | 0 | Deleterious | 1 | Probably Damaging | −8.41 | Deleterious | 0.85 | Probably Damaging | 73 | effect | Disease | 0.86908 | Deleterious |
| rs72481807 | E173K | 0 | Deleterious | 1 | Probably Damaging | −3.25 | Deleterious | 0.78 | Probably Damaging | 84 | effect | Disease | 0.86908 | Deleterious |
| rs72480439 | D291G | 0.015 | Deleterious | 0.985 | Probably Damaging | −6.30 | Deleterious | 0.85 | Probably Damaging | 84 | effect | Disease | 0.86908 | Deleterious |
| rs200182794 | Y349D | 0.022 | Deleterious | 0.917 | Probably Damaging | −6.45 | Deleterious | 0.74 | Probably Damaging | 38 | effect | Disease | 0.86908 | Deleterious |
| rs55771538 | G365R | 0.003 | Deleterious | 0.991 | Probably Damaging | −6.24 | Deleterious | 0.85 | Probably Damaging | 72 | effect | Disease | 0.86908 | Deleterious |
| rs55771538 | G365W | 0 | Deleterious | 0.998 | Probably Damaging | −6.34 | Deleterious | 0.85 | Probably Damaging | 75 | effect | Disease | 0.86908 | Deleterious |
| rs72480442 | R368C | 0 | Deleterious | 0.996 | Probably Damaging | −7.34 | Deleterious | 0.85 | Probably Damaging | 92 | effect | Disease | 0.60548 | Deleterious |
| rs79204362 | R368H | 0.01 | Deleterious | 0.978 | Probably Damaging | −4.59 | Deleterious | 0.85 | Probably Damaging | 90 | effect | Disease | 0.86908 | Deleterious |
| rs104893622 | D374N | 0.006 | Deleterious | 0.981 | Probably Damaging | −4.74 | Deleterious | 0.85 | Probably Damaging | 59 | effect | Disease | 0.71871 | Deleterious |
| rs104893629 | N423Y | 0.001 | Deleterious | 1 | Probably Damaging | −7.62 | Deleterious | 0.85 | Probably Damaging | 89 | effect | Disease | 0.86908 | Deleterious |
| rs201181935 | D430E | 0.002 | Deleterious | 0.922 | Probably Damaging | −3.57 | Deleterious | 0.85 | Probably Damaging | 59 | effect | Disease | 0.86908 | Deleterious |
| rs72481808 | P442A | 0.022 | Deleterious | 1 | Probably Damaging | −7.53 | Deleterious | 0.85 | Probably Damaging | 34 | effect | Disease | 0.86908 | Deleterious |
| rs72549376 | R444Q | 0.003 | Deleterious | 1 | Probably Damaging | −3.77 | Deleterious | 0.85 | Probably Damaging | 69 | effect | Disease | 0.86908 | Deleterious |
| rs148233007 | F445L | 0 | Deleterious | 1 | Probably Damaging | −5.66 | Deleterious | 0.85 | Probably Damaging | 93 | effect | Disease | 0.86908 | Deleterious |
| rs28936701 | R469W | 0 | Deleterious | 1 | Probably Damaging | −7.28 | Deleterious | 0.74 | Probably Damaging | 98 | effect | Disease | 0.60697 | Deleterious |
| rs104894979 | C470Y | 0 | Deleterious | 1 | Probably Damaging | −10.10 | Deleterious | 0.85 | Probably Damaging | 89 | effect | Disease | 0.86908 | Deleterious |
Effect of variations on CYP1B1 protein stability
Two biological tools, namely I-Mutant 2.0 and MUpro, were independently used to investigate the effects of all the 19 missense variations on the CYP1B1 protein stability. The I-Mutant 2.0 server predicted the effect of amino acid change in the form of protein stability Gibb’s free energy change (DDG) and calculated the RI of the variations, while MUpro predicted the effect of protein stability for single site mutation in the form of Gibb’s free energy change (DDG). Of the 19 high-risk missense variations identified, I-Mutant 2.0 revealed that all amino acid variations profoundly affected the CYP1B1 protein by decreasing its stability, except for four variants: G61E, P118L, Y349D, and D430E, which did not influence the protein stability. Apart from I-Mutant 2.0, MUpro identified only P52L had no effect on protein stability and the remaining exhibited a reduction in protein stability. The details of I-Mutant 2.0 and MUpro analysis are provided in Table 3 and Table S2.
Conservancy analysis of high-risk variations and secondary structure of CY1B1
The evolutionary conservation characteristic of all the common 19 missense variants was accomplished by the Consurf server. It is essential to determine the magnitude of an amino acid substitution to alter the protein’s structure and function. Results revealed that all amino acid variations occurred in the highly conserved region of the CYP1B1 protein except for one substitution (Y349D) that was observed in the variable region (Fig. 1). Additionally, the solvent accessibility feature predicted that 13 variants (P52L, G90R, P118L, E173K, D291G, G365W, G365R, R368H, R368C, D374N, D430E, P442A, and R444Q) were found to be highly conserved and exposed residues, four variants (G61E, N423Y, F445L, and C470Y) were observed to be highly conserved and buried residues and two variants (Y349D and R469W) were identified as buried and exposed residues. The evolutionary conservation and solvent accessibility feature of all the 19 substitutions are demonstrated in (Fig. 1). Generally, it has been considered that exposed residues have an impact on protein function and, thereby, highly conserved and exposed residues are recognized to have functional importance. On the other hand, buried residues are incorporated into the protein structure maintenance, and thus highly conserved and buried residues reveal their structural importance (Zhang et al., 2017). Based on this analysis, it can be concluded that changes in 19 regions of the CYP1B1 protein, which play an important role in the structural and functional behavior of protein integrals, confirm their deleterious nature.
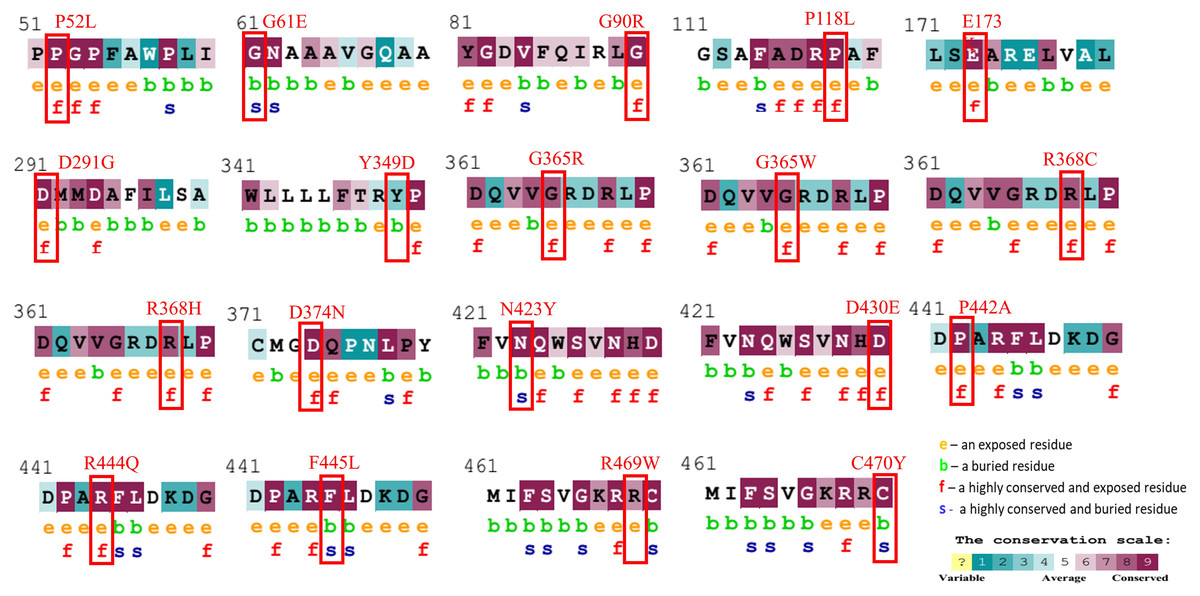
Figure 1: Evolutionary conservancy results of CYP1B1 high-risk missense variants predicted by the ConSurf server.
The red boxes indicate the positions of wild-type amino acids that would be affected by the missense change. In addition, the tool depicted the conservation scores that range from 1 to 9, where conserved sites get scores of 7 to 9, and the characteristics of residue are either exposed (e), buried (b), highly conserved and exposed (functional, f), or highly conserved and buried (s).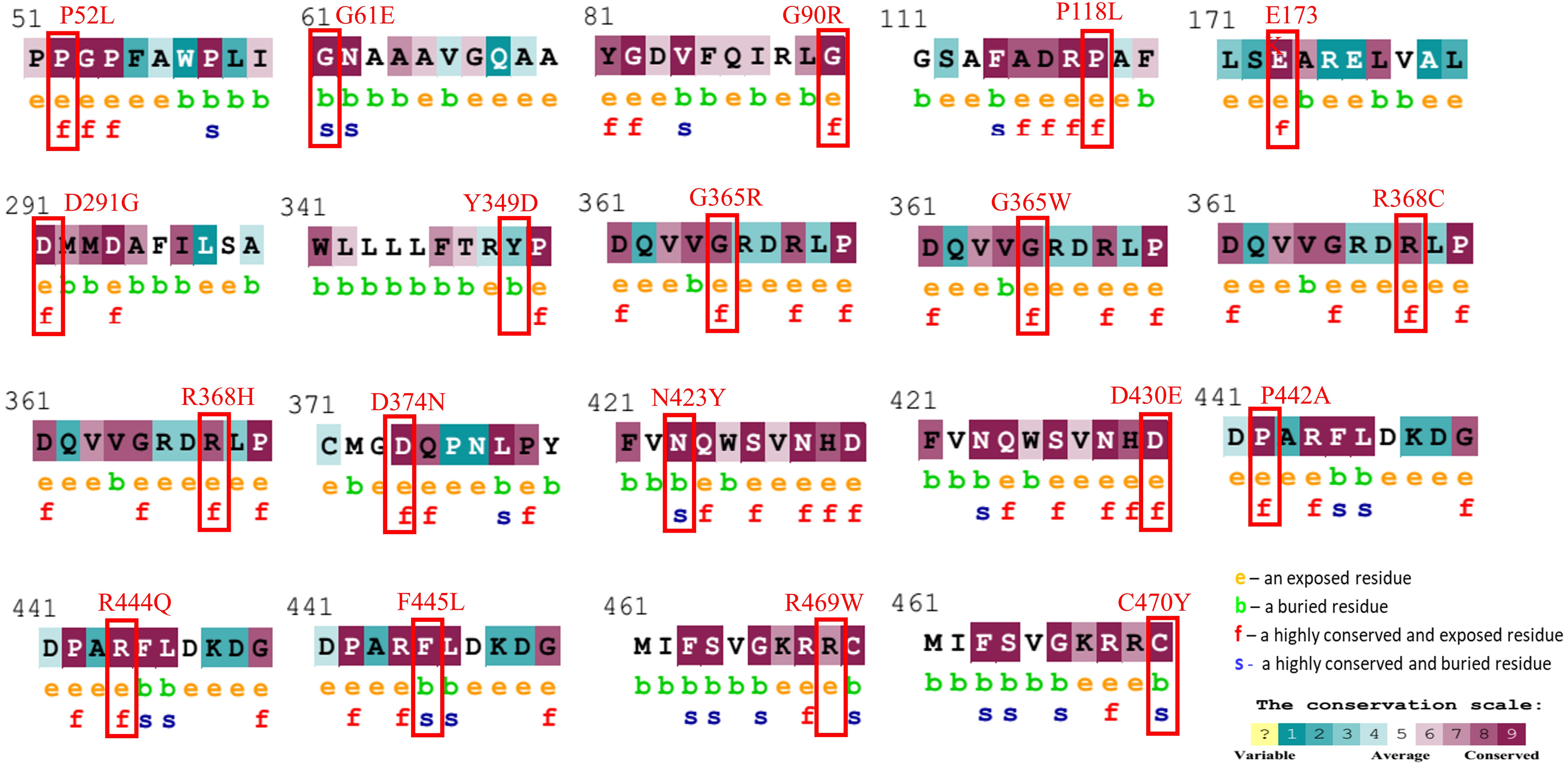{kind=link}
Furthermore, of the total 543 amino acids, 255 amino acids (46.96%) were found in the alpha helix, 207 amino acids (38.12%) in the random coils region, 61 (11.23%) in the extended strand, and 20 (3.68%) in the beta turns of the CYP1B1 protein as predicted by SOPMA (Fig. S1).
Structural examination of native and mutants of CYP1B1
To predict the structural impact of these 19 high-risk variations, first the 3D structure of the CYP1B1 protein was obtained from the PDB database (PDB ID: 3PM0). The structure had some missing residues; therefore, to generate the complete structure of the CYP1B1 protein, the I-TASSER server was utilized. From the generated models of I-TASSER, the best model with a C-score of 0.16, an estimated TM-score of 0.73 ± 0.11, and an estimated RMSD of 7.0 ± 4.1 A was selected and subjected to further evaluation. The quality of the best generated model structure was validated through the ERRAT and VERIFY3D tools of the SAVES server. The ERRAT tool provided the overall quality score, which was 92.57. The VERIFY3D tool estimates the compatibility of a protein model based on local statistical potentials and predicts that the CYP1B1 protein model passed the criteria with an average score of 89.94% and is worth further investigation. Similarly, the 3D structures of all 19 mutants of CYP1B1 were generated, and their graphical illustration and superimposition with the wild-type structure were performed by UCSF Chimera software version 1.14. The 3D wild-type and some mutant structures are depicted in Fig. 2A. In addition, the RMSD of all 19 mutant structures was calculated using the TM-Align server, and their results are shown in Table 3. The results of the TM-Align server elucidated that all 19 mutant structures have profound impacts on the CYP1B1 protein. This suggests that when these variations occur in the CYP1B1 protein, they may interfere with its functional behavior.
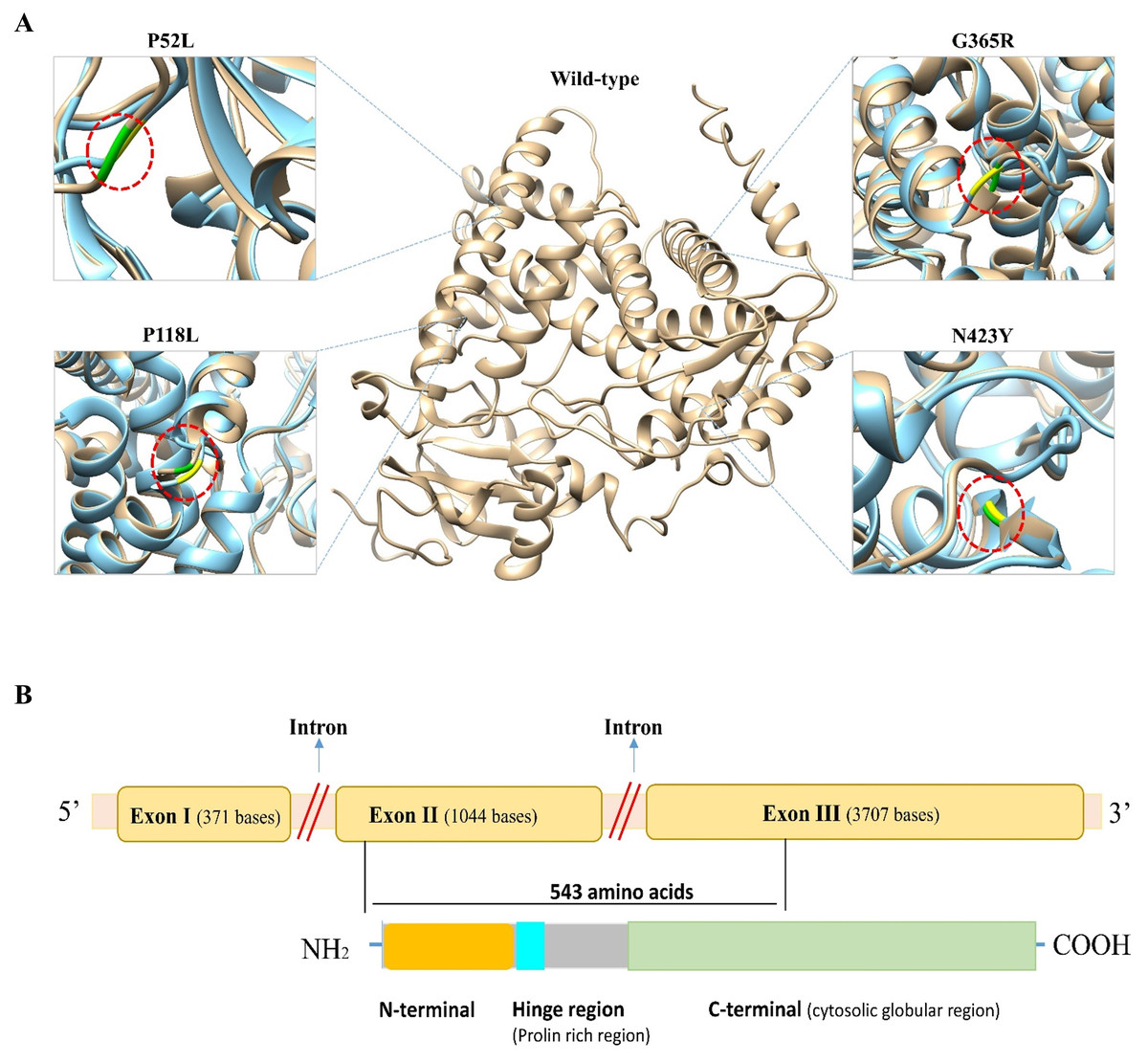
Figure 2: Three-dimensional (3D) structural comparison of wild-type and mutants of CYP1B1 protein.
(A) The wild-type structure is shown in light brown and mutant in light blue colour. The wild-type amino acid is labelled in green, and the mutant substitution is in yellow in the red circular diagrams. (B) A schematic structural diagram of CYP1B1 based on published literature.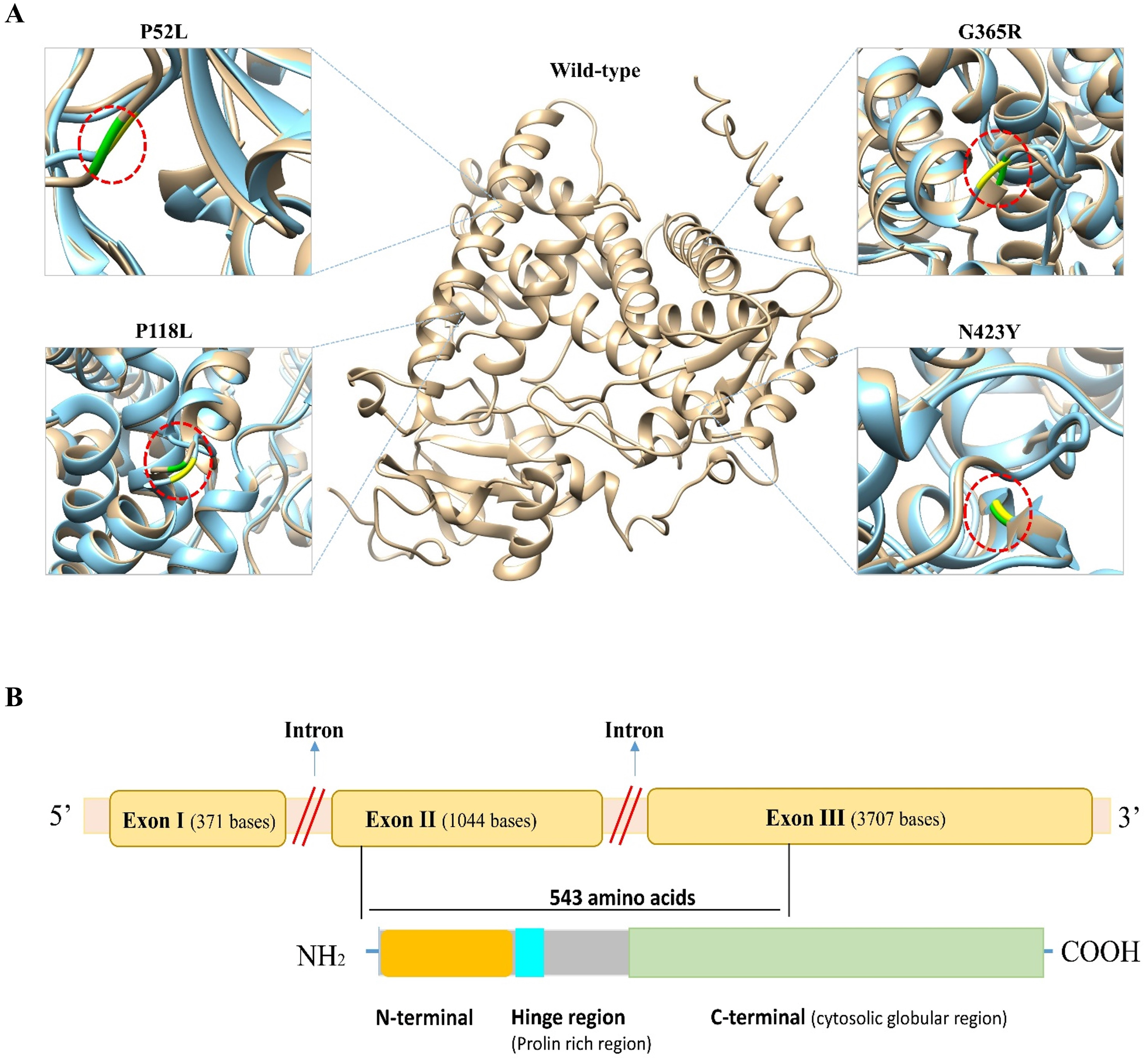{kind=link}
| rs ID | Variant | I-Mutant 2.0 | MUpro | TM Align | gnomAD | Population | |||
|---|---|---|---|---|---|---|---|---|---|
| DDG | Stability | DDG | Stability | RMSD | TM score | Allele frequency | |||
| rs201824781 | P52L | 0.59 | Decrease | 0.470 | Increase | 1.47 | 0.971 | 0.000299/42 | Ashkenazi Jewish, European, Latino, African, others |
| rs28936700 | G61E | −0.2 | Increase | −0.133 | Decrease | 1.2 | 0.984 | 0.000171/24 | European, South Asian, Latino, others |
| rs112014184 | G90R | −1.56 | Decrease | −0.606 | Decrease | 0.98 | 0.989 | Not available | Not available |
| rs73625096 | P118L | 0.68 | Increase | −0.123 | Decrease | 1.53 | 0.978 | 0.000005/1 | Latino |
| rs72481807 | E173K | −0.24 | Decrease | −0.998 | Decrease | 1.3 | 0.980 | 0.000007/1 | Others |
| rs72480439 | D291G | −1.26 | Decrease | −2.096 | Decrease | 1.34 | 0.980 | 0.000008/2 | East Asian |
| rs200182794 | Y349D | −0.03 | Increase | −1.239 | Decrease | 1.26 | 0.985 | 0.000008/2 | African, European (non-Finnish) |
| rs55771538 | G365R | −1.02 | Decrease | −0.502 | Decrease | 1.38 | 0.979 | 0.000004/1 | European (non-Finnish) |
| rs55771538 | G365W | −0.66 | Decrease | −0.606 | Decrease | 1.49 | 0.978 | 0.000004/1 | European (non-Finnish) |
| rs72480442 | R368C | −0.8 | Decrease | −0.59 | Decrease | 1.22 | 0.979 | 0.000207/29 | Latino. European (non-Finnish), others |
| rs79204362 | R368H | −1.53 | Decrease | −0.708 | Decrease | 1.34 | 0.978 | 0.001248/175 | South Asian, East Asian, Ashkenazi Jewish, Latino, European, African |
| rs104893622 | D374N | −0.74 | Decrease | −0.62 | Decrease | 1.27 | 0.979 | Not available | Not available |
| rs104893629 | N423Y | 0.02 | Decrease | −0.750 | Decrease | 1.4 | 0.979 | 0.000007/1 | European (non-Finnish) |
| rs201181935 | D430E | −0.46 | Increase | −0.919 | Decrease | 1.31 | 0.978 | 0.000007/1 | East Asian |
| rs72481808 | P442A | −1.07 | Decrease | −0.72 | Decrease | 0.68 | 0.993 | Not available | Not available |
| rs72549376 | R444Q | −1.39 | Decrease | −1.35 | Decrease | 1.41 | 0.977 | 0.000007/1 | European (non-Finnish) |
| rs148233007 | F445L | −2.41 | Decrease | −0.856 | Decrease | 1.37 | 0.982 | 0.000007/1 | South Asian, European (non-Finnish) |
| rs28936701 | R469W | −0.75 | Decrease | −0.577 | Decrease | 1.38 | 0.977 | 0.000036/5 | South Asian, East Asian, European (non-Finnish), Latino |
| rs104894979 | C470Y | 0.33 | Decrease | −1.111 | Decrease | 1.42 | 0.977 | Not available | Not available |
Molecular dynamics simulation (MDS) analysis
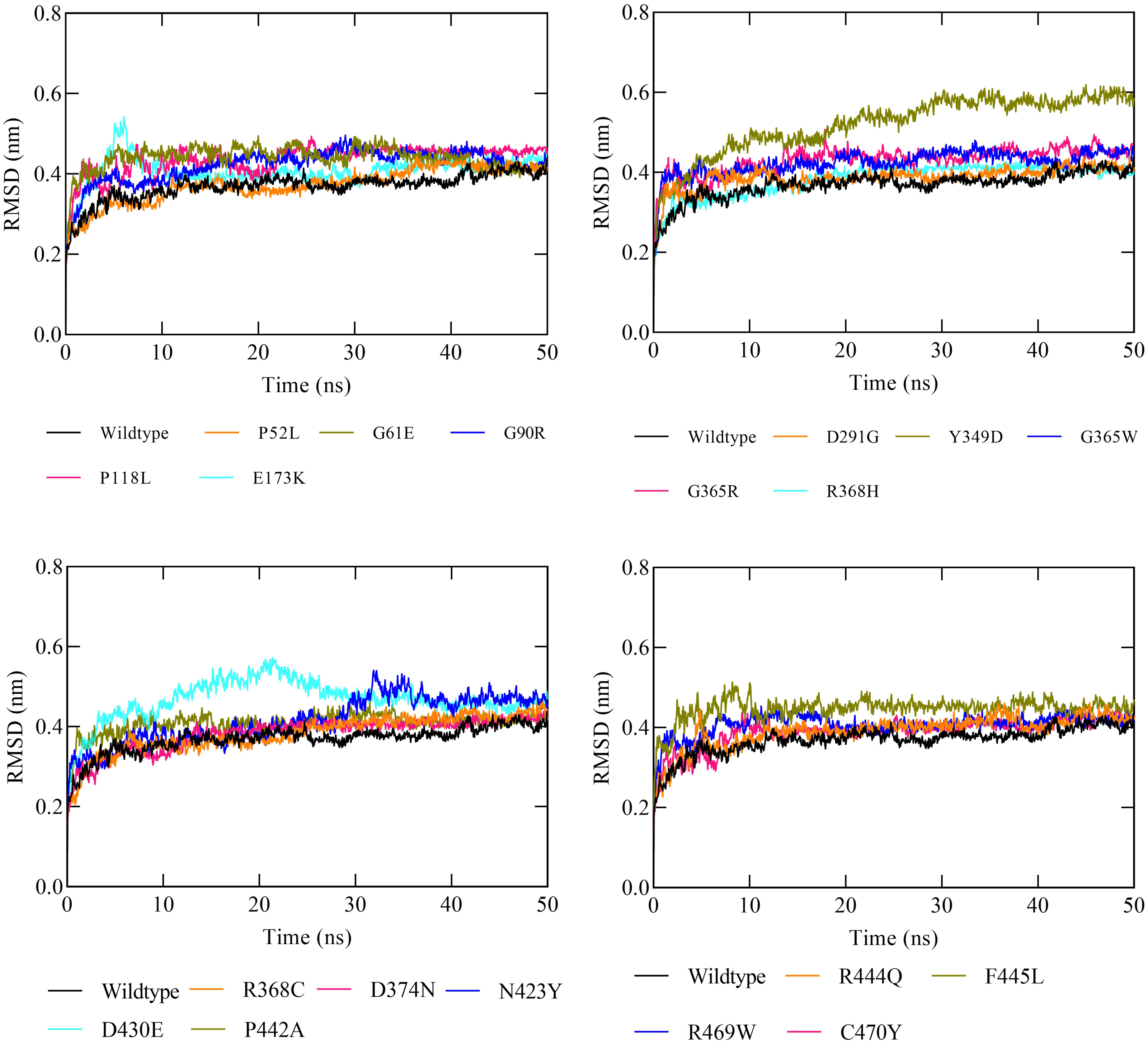
The molecular dynamic simulation (MDS) was executed to evaluate the protein stability following a missense variation in the CYP1B1 protein. The MDS was run up to 50 ns and the RMSD values were noted for the wild-type and for all the 19 high-risk missense variations. Results demonstrated that the CYP1B1 wild-type structure showed an RMDS in the range of 0.36 nm and structural stability throughout the simulation period (Fig. 3). These results indicate that the native structure attained a stable conformation in the MDS period. Overall, the RMSD value was observed to be higher in all the 19 mutants. Six variants (P52L, D291G, R368H, R368C, D374N, and C470Y) demonstrated an RMSD ∼0.38 nm. The highest value of RMSD was observed in three variants (Y349D, D430E, and F445L) that had ∼0.52, 0.46, and 0.44 nm, respectively, compared to the wild-type protein structure. The remaining variants’ RMSD was observed to be greater than 0.40 nm. Figure 3 depicts the RMSD results of all 19 variants after the MDS run. Generally, the higher the RMSD value indicates, the higher the deviation of the mutant with respect to the native protein structure (Agrahari et al., 2019). Therefore, the results of MDS demonstrated that these variants were predicted to destabilize the native protein structure, which could interfere with the normal functioning of the CYP1B1 protein.
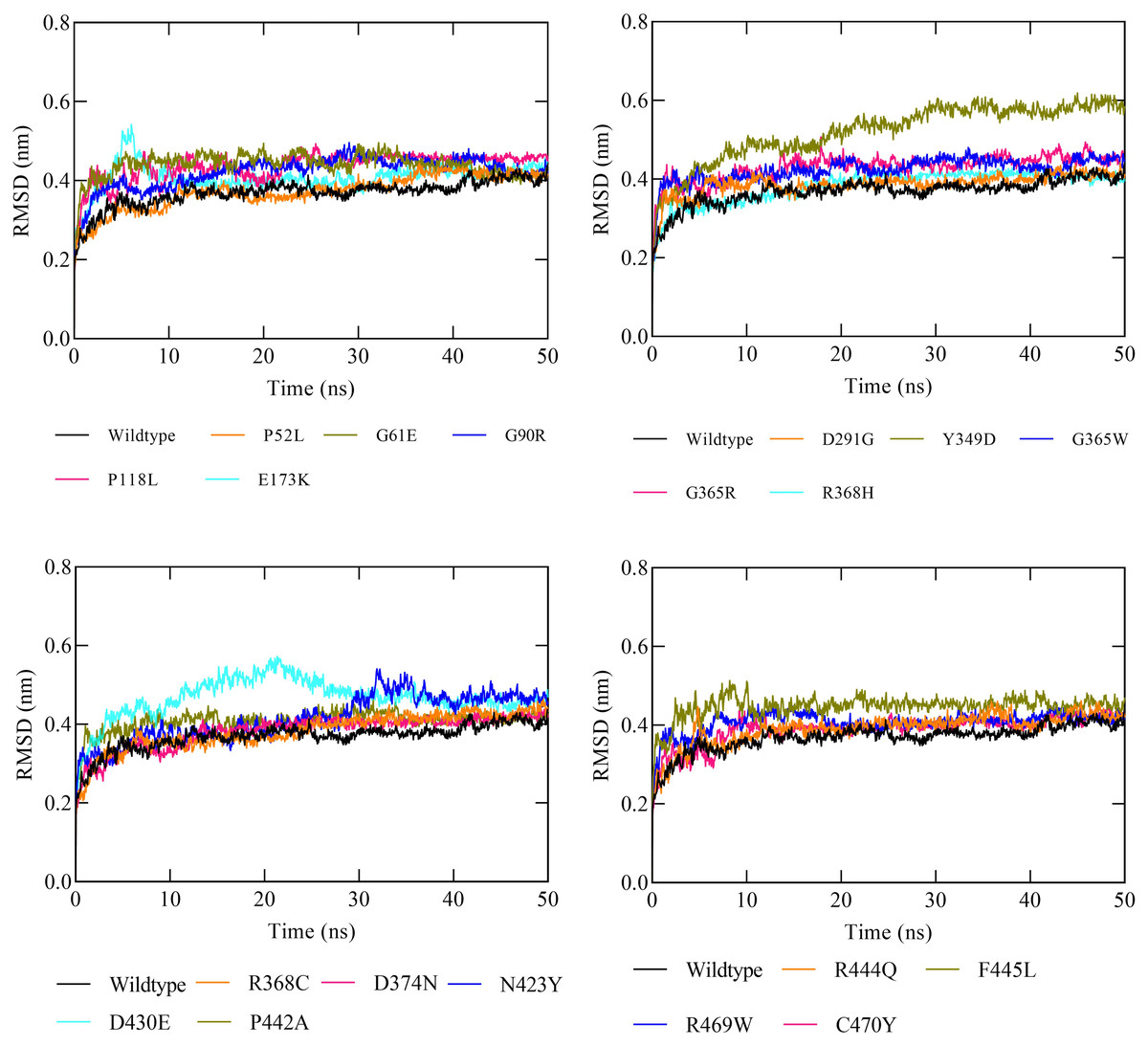
Figure 3: The RMSD results of all the 19 high-risk missense variants of CYP1B1 after the 50 ns MD simulation period.
{kind=link}
Investigation of CYP1B1-interacting candidates
Co-expression data for the CYP1B1 gene was obtained from the GeneMANIA database and then imported into the Cytoscape software to build a network based on the co-expression score (Fig. 4A). Results elucidate that CYP1B1 was highly co-expressed with CYP1A1, followed by PDCD6-AHRR and VCAN genes, respectively. The least expression of CYP1B1 was observed with the LUM, PLA2G7, and F13A1 genes. Furthermore, the protein-protein interaction (PPI) network was also generated via cytoscape software (Fig. 4B). Then, the cytoHubba plugin was employed to identify the highly interacting proteins with CYP1B1. Through cytoHubba, it was discovered that five candidates, which were: CYP1A1, HSD17B1, HSD17B2, AKR1C3, and HSD17B7, demonstrated close interaction with CYP1B1 protein (Fig. 4C).
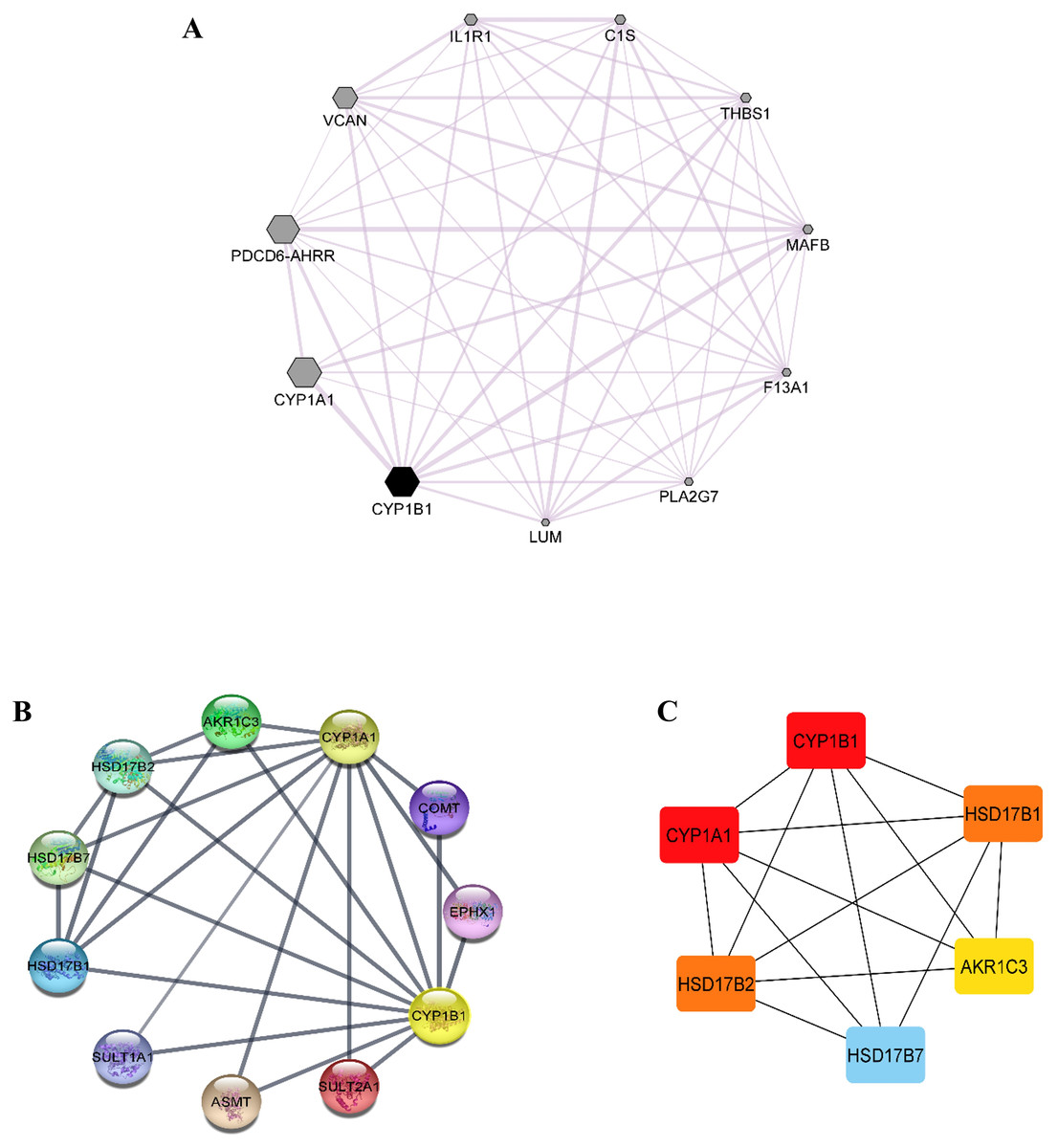
Figure 4: Co-expression and protein-protein interaction (PPI) of CYP1B1.
(A) Co-expression network of CYP1B1 constructed by GeneMANIA plugin of Cytoscape. In the constructed network, the size of the node represents the degree of co-expression. The higher the degree of co-expression, the bigger the size of the node. (B) The protein-protein interaction (PPI) network of CYP1B1. (C) A network of hub candidates predicted to be highly interacted with CYP1B1 by the cytoHubba plugin Cytoscape software.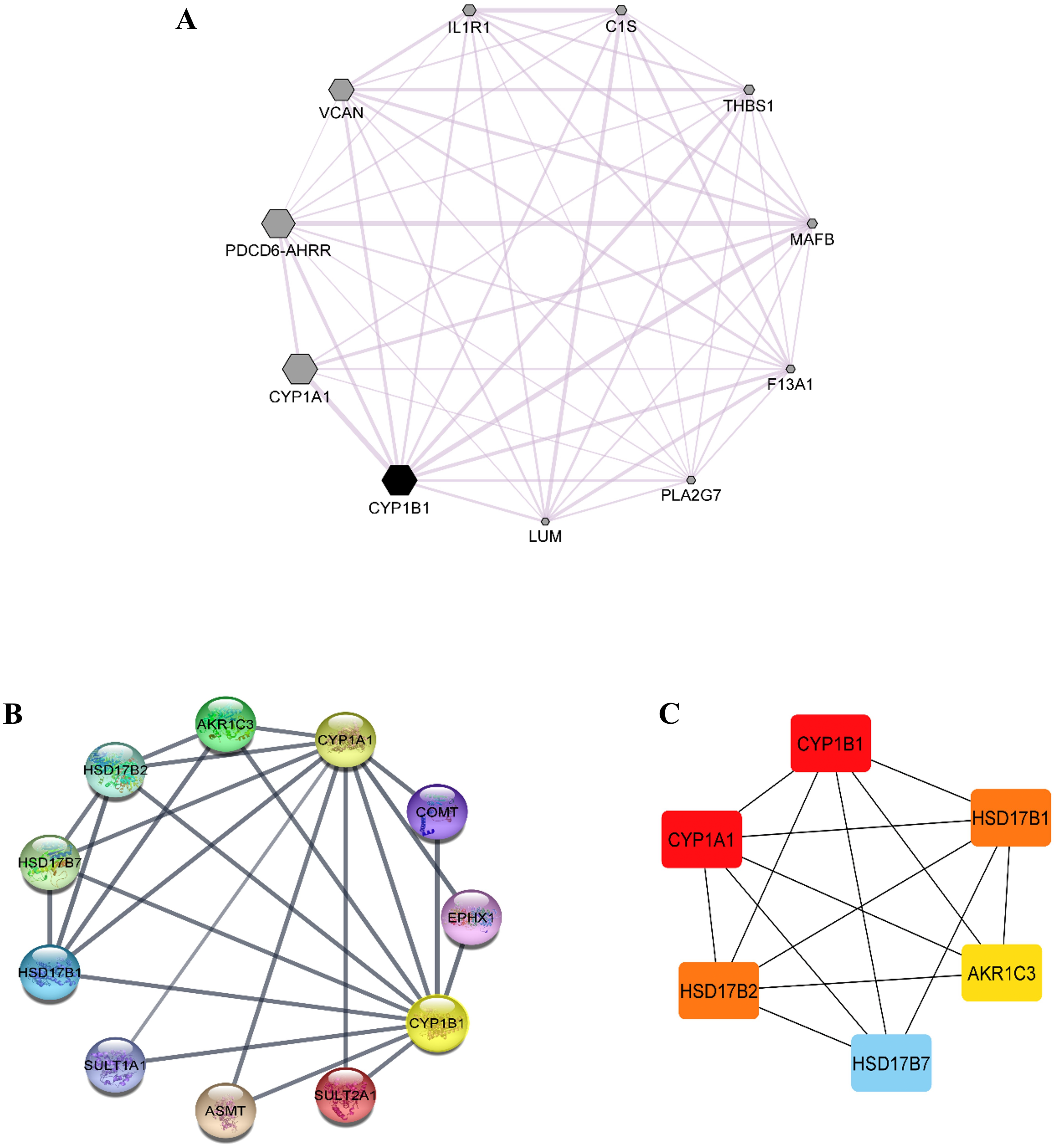{kind=link}
Gene ontology (GO) and pathway enrichment analysis of CYP1B1
In this study, the Enricher server was utilized for Gene Ontology (GO) and pathway analyses. In total, 59 biological processes (BPs) were discovered for CYP1B1 as documented in Table S3. The p-value for all 59 BPs was less than 0.05, indicating that CYP1B1 plays an important biological role in these processes. Fig. 5A illustrates the top ten BPs. Subsequently, only four molecular functions were discovered, which were estrogen 16-alpha-hydroxylase activity, oxidoreductase activity, steroid hydroxylase activity, and heme binding, respectively (Fig. 5B). Likewise, the cellular component (CC) was widely observed in the endoplasmic reticulum membrane and intracellular membrane-bounded organelles. The detailed results of GO are tabulated in Table S3. Moreover, six KEGG pathways were discovered for CYP1B1, which were: Tryptophan metabolism, ovarian steroidogenesis, steroid hormone biosynthesis, metabolism of xenobiotics by cytochrome P450, chemical carcinogenesis, and microRNAs in cancer Fig. 5C Table S4).
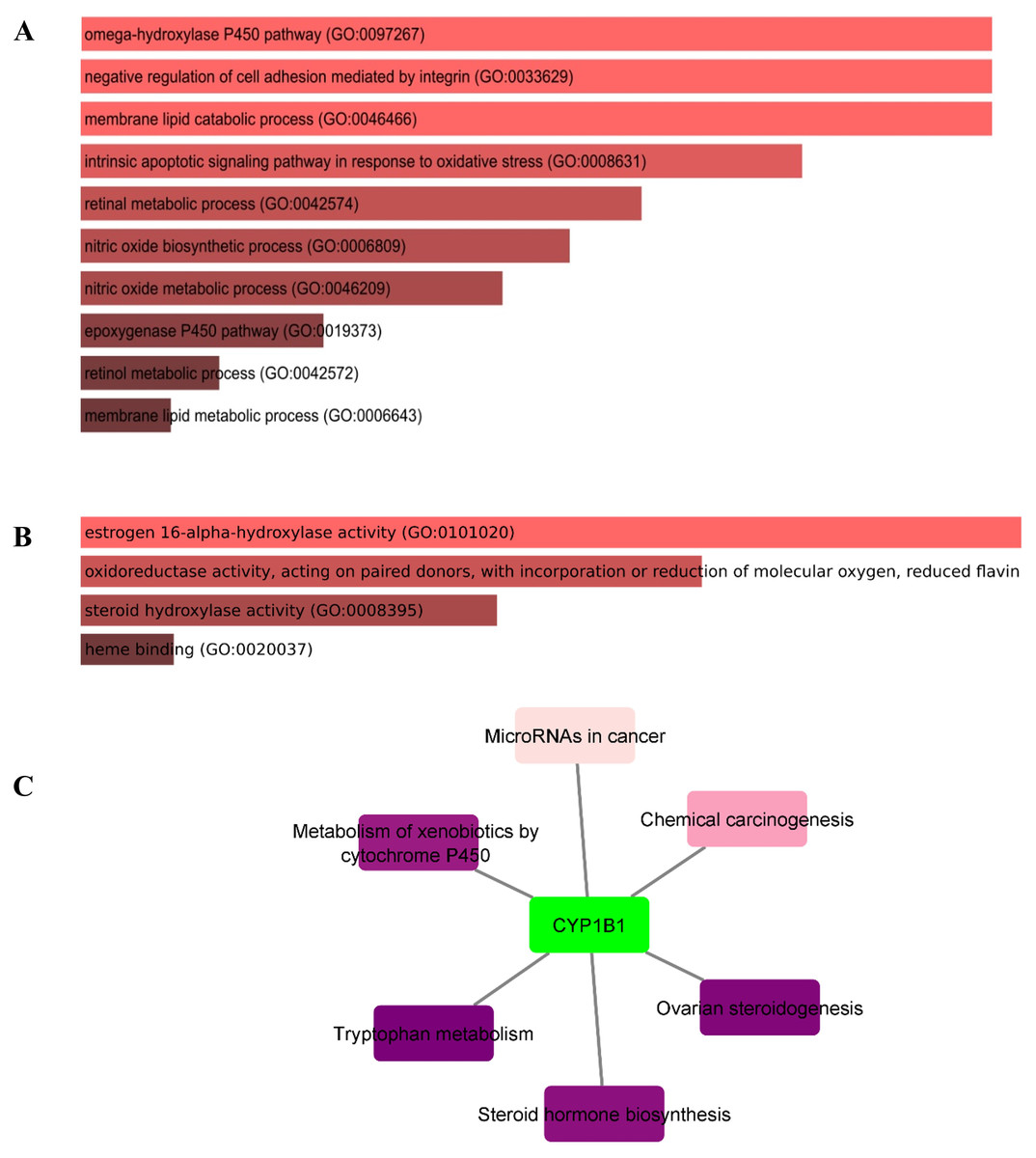
Figure 5: Gene ontology (GO) and pathways analyses of CYP1B1.
The most enriched; (A) biological processes, (B) molecular functions, and (C) KEGG pathways of the CYP1B1.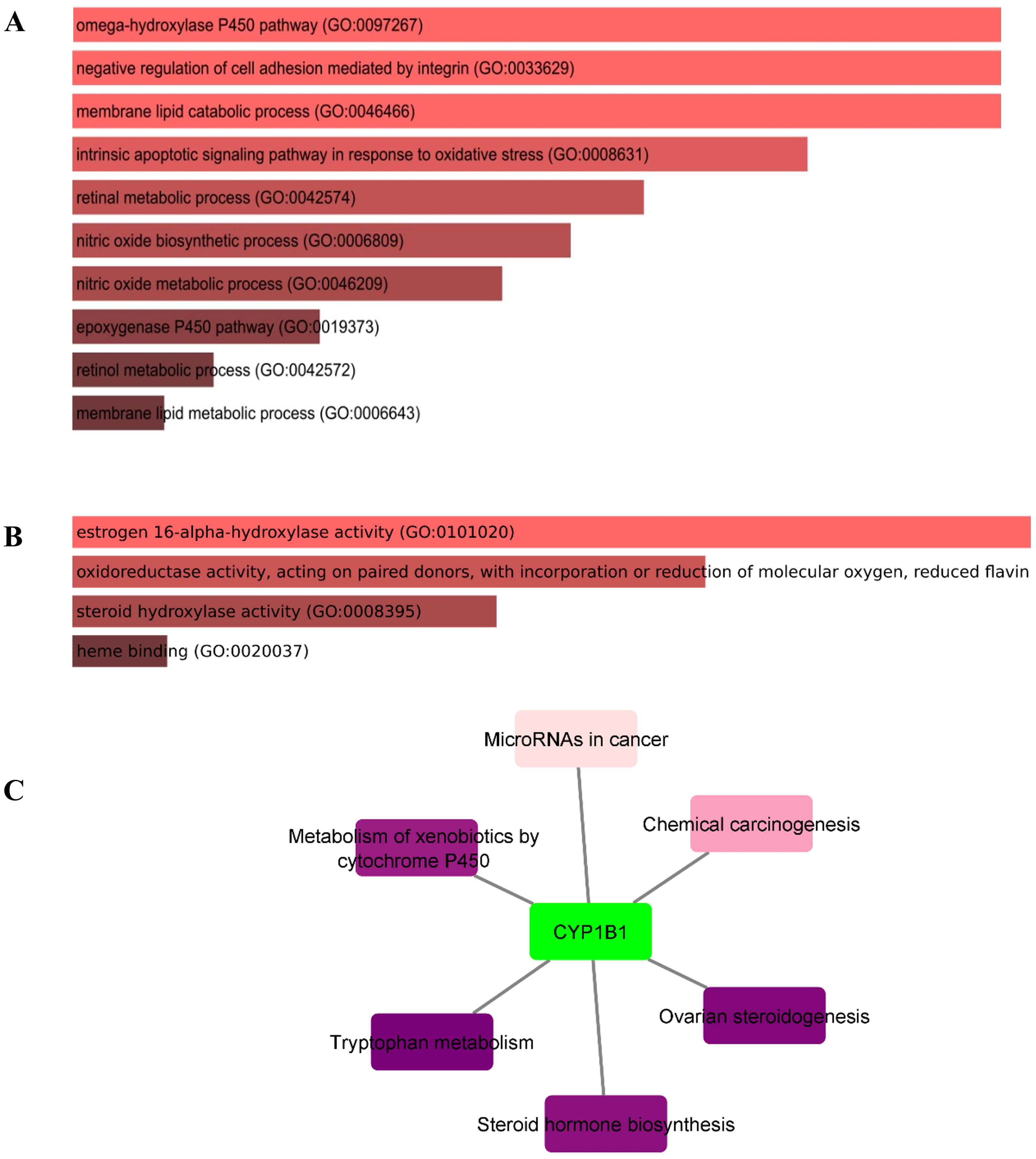{kind=link}
Discussion
The implementation of high-throughput sequencing technology led to the oversaturation of nsSNPs that has recently exponentially increased in human genome databases. In terms of their related contribution to the specific phenotype, characterization of any nsSNP is a cost-effective, cumbersome, and time-consuming procedure, making it hard to evaluate the biological impact of these variations through laboratory trials. In contrast, employing computational techniques assists in filtering out the pathogenic or disease-associated variants from neutrals from a large pool of SNP datasets. Among the two causative genes (CYP1B1 and LTBP2) of primary congenital glaucoma (PCG), CYP1B1 is found to be highly mutated and responsible for the etiology of PCG patients worldwide (Firasat et al., 2008). Therefore, a comprehensive systematic approach was carried out to evaluate the impacts of missense variations at functional and structural levels.
After employing comprehensive functional analysis, 19 missense variants (P52L, G61E, G90R, P118L, E173K, D291G, Y349D, G365W, G365R, R368H, R368C, D374N, N423Y, D430E, P442A, R444Q, F445L, R469W, and C470Y) were found to be common and were considered the high-risk deleterious variants of the CYP1B1 gene (Table 2). At position 52, the proline amino acid residue was replaced by leucine, a bigger residue than the wild-type. Prolines are known to have very rigid structures, and any substitution with proline could disturb the local structure of the protein. This variant was previously discovered by Campos-Mollo et al. (2009). They carried out a functional analysis study by transiently transfected human embryonic kidney 293T (HEK-293-T) cells and found that the P52L variant reduced the catalytic activity of CYP1B1 and imparted high protein instability (Campos-Mollo et al., 2009). Conservancy analysis revealed that this variation occurred in the highly conserved and exposed region of the protein, highlighting its magnitude of penetrance in malfunctioning of the protein (Fig. 1). The stability and MDS analysis also demonstrated the role in the reduction of protein stability and RMSD deviation (0.37 nm) of this variant protein compared with the native protein (Fig. 3). Likewise, at position 61, the hinge region, where glycine, which is a neutral amino acid, was substituted by a negatively charged glutamate. This variant (G61E) was identified in many populations, as reported in previous studies (Afzal et al., 2019; El-Ashry, El-Aziz & Bhattacharya, 2007; El-Gayar et al., 2009; Jubair et al., 2019; Stoilov et al., 1998). In this study, it has been shown that most of the high-risk missense variations are found in the carboxyl terminal region of the CYP1B1 protein, which is incorporated into several catabolic activities. This variation was found to be in the highly conserved and buried region and showed a high RMSD deviation value (0.43 nm) compared to the wild-type protein (Fig. 3). The mutated residue introduced a negative charge, which may cause repulsion of other residues with the same charge and may interfere with the protein’s functional activity or incorrect conformation of the protein structure.
Two variants (G90R and G365R) where glycine, a neutral amino acid, was mutated by a positively charged arginine, exhibited instability as well as high RMSD (0.42 and 0.43 nm, respectively), which could affect the proper protein folding (Fig. 3). Whereas, three variants (R368C, R444Q, and R469W) in which a positively charged amino acid (Arginine) was replaced by three neutral amino acids (Cysteine, Glutamine, and Tryptophan) at positions 368, 444, and 469, respectively. The substituted residues are bigger in size and change the charge of the native residue, which may lead to protein folding problems and will disturb the local structure (Shahid et al., 2022). R368C and R444Q existed in the highly conserved and exposed region, and R469W was in the buried region of the protein, influencing the reduction in stability and showing an RMSD of around 0.40 nm (Figs. 1 and 3). The frequency of these three variants is very high, as they were identified in various populations (Table 3) (Faiq et al., 2013; Li et al., 2011; Stoilov et al., 1998). Similarly, the remaining high-risk missense variations (P118L, E173K, D291G, Y349D, G365W, R368H, D374N, N423Y, D430E, P442A, F445L, and C470Y) were found to be distributed in many populations as discovered by various research studies (Bejjani et al., 2000; Chen et al., 2015; Colomb, Kaplan & Garchon, 2003; El-Gayar et al., 2009; Li et al., 2011; Stoilov et al., 1998). The findings suggest that these variants may influence the protein structure and functions, either by misfolding the local structure or resulting in the loss of protein–protein interactions of the CYP1B1 protein. Generally, the higher the RMSD value indicates, the higher the deviation of the mutant with respect to the native protein structure (Agrahari et al., 2019). Therefore, the results of MDS demonstrated that these variants were predicted to destabilize the native protein structure, which could interfere with the normal functioning of the CYP1B1 protein.
In humans, the CYP1B1 gene comprises three exons with 371, 1,044 and 3,707 base pairs in length, respectively, that encode a protein with a length of 543 amino acids. It has been seen that the coding region for the CYP1B1 protein starts from the 5′ end of exon number two and ends within the third exon (Faiq et al., 2013). This protein has three structural regions: N-terminal membrane-bound region of 53 residues; a hinge region comprised of 10 proline-rich amino acid residues that participate in the flexibility of the protein; and a 480 amino acid long C-terminal harboring a heme-binding region (Vasiliou & Gonzalez, 2008). The C-terminal is a highly conserved cytosolic globular domain that is essential for the normal function of this class of enzymes. A schematic diagram of CYP1B1 based on a literature review has been shown in Fig. 2B.
Co-expression analysis demonstrated that CYP1B1 was highly co-expressed with CYP1A1, followed by PDCD6-AHRR and VCAN genes, respectively (Fig. 4A). The CYP1A1 also belongs to the same superfamily of cytochrome P450 and its encoded product performs similar functions as the CYP1B1, i.e., detoxification of xenobiotics, drug metabolism, synthesis of cholesterol, steroids, and other lipids (Kyoreva et al., 2021). The VCAN is a member of the versican proteoglycan family. The encoded product is incorporated into cell proliferation, migration, adhesion, and angiogenesis. It also plays a central role in tissue morphogenesis and maintenance (Wight, 2002). However, these results delineate the additional functions of the CYP1B1 gene by being co-expressed with these genes. The PPI network analysis discovered that five candidates, which were: CYP1A1, HSD17B1, HSD17B2, AKR1C3, and HSD17B7, exhibited close interaction with CYP1B1 protein (Figs. 4B and 4C). The CYP1A1 protein is a member of the CYP protein family, which plays a significant role in the activation of compounds with carcinogenic properties (30). AKR1C3 (Aldo-keto reductase family 1 member C3) is an enzyme that catalyses the conversion of aldehydes and ketones into their respective alcohols. This enzyme is one of the promising biomarkers for the development of prostate cancer (29). HSD17B1, HSD17B2, and HSD17B7 are the members of the 17 β-hydroxysteroid dehydrogenase family that are incorporated in the end phase of the biosynthesis of active steroid hormones. HSD17B1 and HSD17B7 are reductive enzymatic proteins, while HSD17B2 is an oxidative enzymatic protein (Lukacik, Kavanagh & Oppermann, 2006). The findings of this study suggest that CYP1B1 might have many functions by interacting with CYP1A1, HSD17B1, HSD17B2, AKR1C3, and HSD17B7 proteins.
The GO analysis revealed that CYP1B1 is predominantly involved in the omega-hydroxylase P450 pathway, negative regulation of cell adhesion mediated by integrin, membrane lipid catabolic process, intrinsic apoptotic signaling pathway in response to oxidative stress, retinal metabolic process, and many other biological processes (Table S3). Figure 5A illustrates the top ten BPs. CYP1B1 was discovered to have six KEGG pathways, which were: Tryptophan metabolism, ovarian steroidogenesis, steroid hormone biosynthesis, metabolism of xenobiotics by cytochrome P450, chemical carcinogenesis, and microRNAs in cancer (Fig. 5C). All these findings were consistent with the earlier research studies and suggest that CYP1B1 is a multifunctional protein and is incorporated into the regulation of multiple pathways.
However, there were several admitted confines to this study, such as that it was an in silico study where publically available data was utilized for the analysis. We employed a comparative approach to improve the accuracy and enhance the reliability, but there were some pitfalls in the study as these were the computational tools that used different algorithms. These tools possibly provide some false negative or positive outcomes due to overlapping concepts in their predictions that restrict us from being fully confident unless verified through wet lab experiments. Moreover, although most of the predicted variants have been reported in various research studies, experimental validation is still required to affirm the actual consequences. Functional assays, segregation analysis, genetic studies, and epidemiological data could be implemented to predict the pathogenicity and improve the classification accuracy, respectively. Despite these limitations, the study presented here to some extent overcomes them as it was a comprehensive in silico study where molecular dynamic simulation was also performed to enhance the reliability of the results, thereby providing a quick, critical, and cost-effective approach to evaluate the mutational spectra of the CYP1B1 gene and to discover the probable biomarkers that can be helpful for the understanding and management of primary congenital glaucoma.
Conclusion
The present study was focused on identifying the high-risk missense deleterious variants of the CYP1B1 gene that are associated with primary congenital glaucoma (PCG) through comparative in silico approaches. A spectra of 19 high-risk missense deleterious variations, which were primarily involved in PCG pathogenesis, were identified from all of the retrieved data. Furthermore, phylogenetic analysis, molecular dynamic simulation results, and structural analysis of the wild-type and mutant forms revealed that the majority of the variants were in highly conserved regions and could reduce protein stability, potentially causing CYP1B1 protein dysfunction. Moreover, the results of the gene expression, protein-protein interaction, gene ontology, and pathway analysis were mostly consistent with the earlier research studies. Nevertheless, this study provides an essential profile of CYP1B1 and its highly associated missense variants with the PCG that can be potentially used as diagnostic markers and will be helpful for the management of primary congenital glaucoma studies.
Supplemental Information
CYP1B1 protein stability results derived from I-Mutant and MUpro web tools
Molecular Dynamics Simulation raw data (replicate 1)
The XVG files can be opened by using Gromacs Software plugins (https://www.gromacs.org/).
Molecular Dynamics Simulation Raw data (replicate 2)
The XVG files can be opened by using Gromacs Software plugins (https://www.gromacs.org/).