Photoperiod induced the pituitary differential regulation of lncRNAs and mRNAs related to reproduction in sheep
- Published
- Accepted
- Received
- Academic Editor
- Mohammed Gagaoua
- Subject Areas
- Agricultural Science, Genetics, Genomics, Molecular Biology, Zoology
- Keywords
- Sheep, Pituitary, RNA sequencing, Photoperiod, lncRNA, mRNA
- Copyright
- © 2021 He et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2021. Photoperiod induced the pituitary differential regulation of lncRNAs and mRNAs related to reproduction in sheep. PeerJ 9:e10953 https://doi.org/10.7717/peerj.10953
Abstract
The pituitary is a vital endocrine organ that regulates animal seasonal reproduction by controlling the synthesis and secretion of the hormone. The change of photoperiod is the key factor affecting the function of the pituitary in animals, but the mechanism is unclear. Here, we studied the transcriptomic variation in pars distalis (PD) of the pituitary between short photoperiod (SP) and long photoperiod (LP) using RNA sequencing based on the OVX+E2 sheep. 346 differentially expressed (DE) lncRNAs and 186 DE-mRNA were found in the PD. Moreover, function annotation analysis indicated that the reproductive hormones and photoperiod response-related pathways including aldosterone synthesis and secretion, insulin secretion, thyroid hormone synthesis, and circadian entrainment were enriched. The interaction analysis of mRNA-lncRNA suggested that MSTRG.240648, MSTRG.85500, MSTRG.32448, and MSTRG.304959 targeted CREB3L1 and DUSP6, which may be involved in the photoperiodic regulation of the PD. These findings provide resources for further study on the seasonal reproductive in ewes.
Introduction
Animals can schedule reproduction events to maximize adaptation to the changing environment and the survival of offspring. Most animals, including birds, mammals and even in the human, have a highly accurate mechanism for photoperiod measurement and show dramatic changes in seasonal response to small changes in photoperiod (Nakayama & Yoshimura, 2017; Guh, Tamai & Yoshimura, 2019). In sheep, the efficiency of reproduction is significantly related to the frequency of estrus (Li et al., 2019a). However, photoperiodic seasonal change is the main inducement that affects female mammalian seasonal estrus and reproduction. Compared to ewes within estrous cycling (short photoperiod, SP) with anestrus (long photoperiod, LP), early studies showed that there are significant physiological and neuroendocrine differences in pituitary (Mcbride et al., 2012). It is well known that the start and stop of mammalian reproductive activities are controlled by hypothalamic-pituitary-gonadal axis (HPGA), and several reproductive hormones such as follicle-stimulating hormone (FSH), luteinizing hormone (LH), prolactin (PRL) are all secreted by PD of the pituitary gland (Nestor et al., 2018; Lincoln et al., 2006; Dardente et al., 2019), importantly, these hormones vary with the seasons or photoperiod in birds, goats and sheep (Nakayama & Yoshimura, 2017; Guh, Tamai & Yoshimura, 2019; Di et al., 2014; Zhai et al., 2018; Li et al., 2019a).
Remarkably, most of the reproductive hormones are proteins, non-coding RNA should be taken into account transcriptional regulation of their synthesis and secretion. LncRNA play a vital role in regulating the mammalian reproduction by involving in a series of biological processes such as gametogenesis (Mercer et al., 2010; Arun et al., 2012), placentation (Gao et al., 2012), reproductive hormone responses (Gao et al., 2012; Liu et al., 2018a). In our recent studies, we found that several differentially expressed lncRNAs (DE-lncRNA) in the hypothalamus and uterus may be involved in the sheep reproductive process by interacting with target genes in Small Tail Han sheep (Lai et al., 2012; Zhang et al., 2019). Moreover, Zheng et al. (2019) found that the target genes of the DE lncRNAs were significantly enriched in pituitary function and hormone-related pathways, which may participate in the ovine prolificacy. However, the studies on the expression pattern and potential roles of lncRNAs in the pituitary are still limited compared to the miRNAs, lncRNAs or mRNAs. The pituitary is an important endocrine gland that plays a connecting role in the hypothalamus and gonad. However, the work about the systematic analysis of expression pattern of lncRNAs in the key area of pituitary gland (such as PD) during the different photoperiod has not been performed.
To date, our understanding of photoperiod induced molecular neuroendocrine is still limited. In this study, to identify the role of lncRNAs and mRNAs in the pituitary associated with sheep reproduction based on different photoperiod treatments, high-throughput sequencing was performed on the PD of pitiutary to screen DE genes and DE lncRNAs, subsequently, bioinformatics analysis was used to identify the key lncRNAs that regulate the pituitary function and photoperiodic response in the PD. This study expands the understanding and catalogue of PD lncRNA in sheep, and provides candidate regulators of sheep reproduction regulation at the transcriptional level.
Material and Methods
Animal treatments and sample collection
The six Sunite ewes (35–40 kg, 3 years-old, clinically normal and non-pregnant) were selected and housed in the Tianjin Institute of Animal Sciences, Tianjin (39°N latitude), China. All ewes were raised under the same conditions, fed ad libitum and had free access to water. Before the experiment, the 6 Sunite ewes were ovariectomized (OVX) via midventral laparotomy the surgical procedures are as follows: firstly, we fixed the sheep on the frame to make the breasts fully exposed, and washed the stains around the abdomen with hot soapy water, then we scraped the wool cleanly. The surgical instruments and the hands of the surgeon were routinely disinfected. After the surgery, the ewes were allowed to recover for at least 30 days before hormone treatments. 30 days after arrival, 6 ewes were treated with Estradiol as described in the previous studies (Smith et al., 2007). Finally, 6 ewes were maintained in two photoperiod-controlled room (Room1: SP, Short Photoperiod, 8/16 h light-black; Room2: LP, Long Photoperiod, 8/16 h light-black). The slaughter was scheduled for SP42 day and LP42 day, Pituitary was rapidly removed from the brain, the PD was quickly separated,frozen in liquid nitrogen, and stored at −80 °C for subsequent study.
All the experimental procedures were approved by the Science Research Department of the Institute of Animal Sciences, Chinese Academy of Agricultural Sciences (IAS-CAAS) (Beijing, China). And ethical approval was given by the Animal Ethics Committee of the IAS (IAS2018-3).
RNA Extraction and sequencing
Total RNA from each sample was isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA purity, concentration, and integrity were also detected following our previous method (La et al., 2019). Subsequently, sequencing libraries were generated using the rRNA-depleted RNA by NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA). Finally, the libraries were sequenced on an Illumina platform.
Sequence analysis
Ovis aries reference genome and gene annotation files were downloaded from the genome website directly (Oar_v4.0, https://www.ncbi.nlm.nih.gov/assembly/GCF_000298735.2). Clean data were obtained by removing reads containing adapter, ploy-N, and low quality reads were also removed from raw data. HISAT2 was used to map the clean reads to the reference genome, the software was run with ‘–rna-strandness RF’, and other parameters were set as default. The mapped reads of each sample were assembled by StringTie following the reference-based approach (Pertea et al., 2016). The assembled transcripts were annotated using the gffcompare program. Transcripts with length more than 200 nt and two exons were selected as lncRNA candidates. CPC, CNCI, Pfam, CPAT were used to distinguish the protein-coding genes from the non-coding RNAs. After the above four analyses, the remaining lncRNAs were used for subsequent analysis.
Differential expression genes identification and qPCR validation
FPKM was used to measure the expression levels of transcripts. HTSeq was used to calculate the FPKM of both lncRNAs and mRNAs in each sample based on the Python, the program was run with ‘-igene_id -f bam –s reverse -a 10 –q’. A criterion of absolute log2 (fold change)>1 and P < 0.01 was used to identify differentially expressed genes using‘MARS’ of the Deseq. RNA sequencing results were validated by qPCR, in which 5 mRNAs and 5 lncRNAs were randomly selected, the information of primers were listed in Table 1. The cDNA synthesis and qPCR was performed as described in our previous study (Zhang et al., 2019). All samples were examined in triplicate. Relative expression levels of differentially expressed mRNAs and lncRNAs were normalized by β-actin using 2 −ΔΔCt method (Livak & Schmittgen, 2000).
| Transcript type | Transcript name | Forward primer | Reverse primer | Product size (bp) |
|---|---|---|---|---|
| mRNA | DUSP6 | CTGGAACGAGAATACTGGCG | ATCTTCCAGGTAGAACGCCC | 91 |
| ANGPTL2 | AACTGTGCCCACTACCAGAA | ATCACCACCTTCTTGAGCGA | 164 | |
| SPRY4 | CGTTGGTGCAGTGGTAGAAG | CGTTGGTGCAGTGGTAGAAG | 152 | |
| GHRH | CAGCAGGGAGAGAGAAACCA | CCAAGATGCTCTCCAATGCC | 106 | |
| FOSL2 | TGCAGAAGGAGATCGCTGAA | CAGGACTGATCTTGCACACG | 90 | |
| LncRNA | MSTRG.251736 | ATCAGCATTCCCCTCTCTGG | CACAGACTGTTCTTTGCCCC | 147 |
| MSTRG.202202 | ATCAGCTCTCCCAGTCACAC | GCCTCCCTCTCTGAAAACCT | 136 | |
| MSTRG.26775 | ACAAGGAGCTGATGTCACCA | TCAGGGTGGAGAGTGAGGTA | 155 | |
| MSTRG.130282 | TGGTTTTCATTCTGCCTGCC | GATTTACTGTGCATGGCCCC | 127 | |
| MSTRG.50976 | AACCCTAACCAGAGATGGCC | TCTAGAGGCTGTTTCTGGGC | 93 |
Functional annotation and enrichment analysis
The cis and trans functions of lncRNA were defined in our previous study (Zhang et al., 2019). The R packages, such as KOBAS and Goseq, were used to implement GO and KEGG enrichment analysis (Zheng et al., 2019). a corrected P < 0.05 (q < 0.05) were considered significantly enriched for GO and KEGG enrichment.
Construction of mRNA–mRNA and lncRNA–mRNA networks
We select the mRNAs with High Spearman correlation Coefficient (P ≥ 0.9) as the trans-targets of DE-lncRNA, and the mRNA with distance less than 50 kb were selected as the Cis-targets of DE-lncRNA. To further explore the interactions between the DE-lncRNAs, target genes and DE-mRNAs in PD, The interaction networks of transcripts associated with pituitary function and reproduction were built using the DE-lncRNA, target genes of lncRNAs and DE-mRNAs. Visualization was achieved through software called Cytoscape (V3.6.1) (La et al., 2019; Zheng et al., 2019; Zhang et al., 2019).
Statistical analysis
Student’s T-test of SPSS 20.0 statistical software were used to evaluate the experimental results . All data were shown as means with standard error (SE). one-way ANOVA was used for qPCR validation. P < 0.05 was considered statistically significant.
Results
Summary of sequencing data in ovine PD
In this study, 6 libraries were established to identify the changes in sheep PD transcriptome in both SP and LP. A total of 94.48G clean bases were obtained. The percentage of clean Q30 is more than 92.64%, and the ratio of clean reads and total mapped reads was larger than 95.29% and 93.57% respectively (Table 2).
| Sample | Raw reads number | Clean reads number | Clean reads rate (%) | Clean bases (G) | Clean Q30 bases rate (%) | Total mapping rate (%) |
|---|---|---|---|---|---|---|
| SP42P1 | 116,206,488 | 113,856,274 | 97.98 | 15.91 | 93.79 | 94.29 |
| SP42P2 | 117,226,392 | 112,967,194 | 96.37 | 15.78 | 92.64 | 93.57 |
| SP42P3 | 129,006,964 | 125,514,778 | 97.29 | 17.53 | 94.03 | 94.23 |
| LP42P1 | 111,936,010 | 107,803,564 | 96.31 | 15.06 | 94.11 | 95.21 |
| LP42P2 | 116,237,926 | 110,762,742 | 95.29 | 15.47 | 94.02 | 93.39 |
| LP42P3 | 106,513,748 | 103,666,030 | 97.33 | 14.48 | 93.99 | 94.12 |
Identification of lncRNAs and mRNAs in ovine PD
After mapping to the sheep genome (Oar_v4.0), 48308 novel lncRNA were identified (Fig. 1A), in which the maximum proportion of intronic lncRNAs was 56.94%, followed by lincRNAs and antisense lncRNAs for a minimum percentage (Fig. 1B). The expression of transcripts in SP is higher than LP (Fig. 1C). Moreover, length between lncRNA and mRNA is similar in the PD (Figs. 1D–1E), the exon number of lncRNA is less than mRNA and most of which have 2 or 3 exons (Figs. 1F–1G).
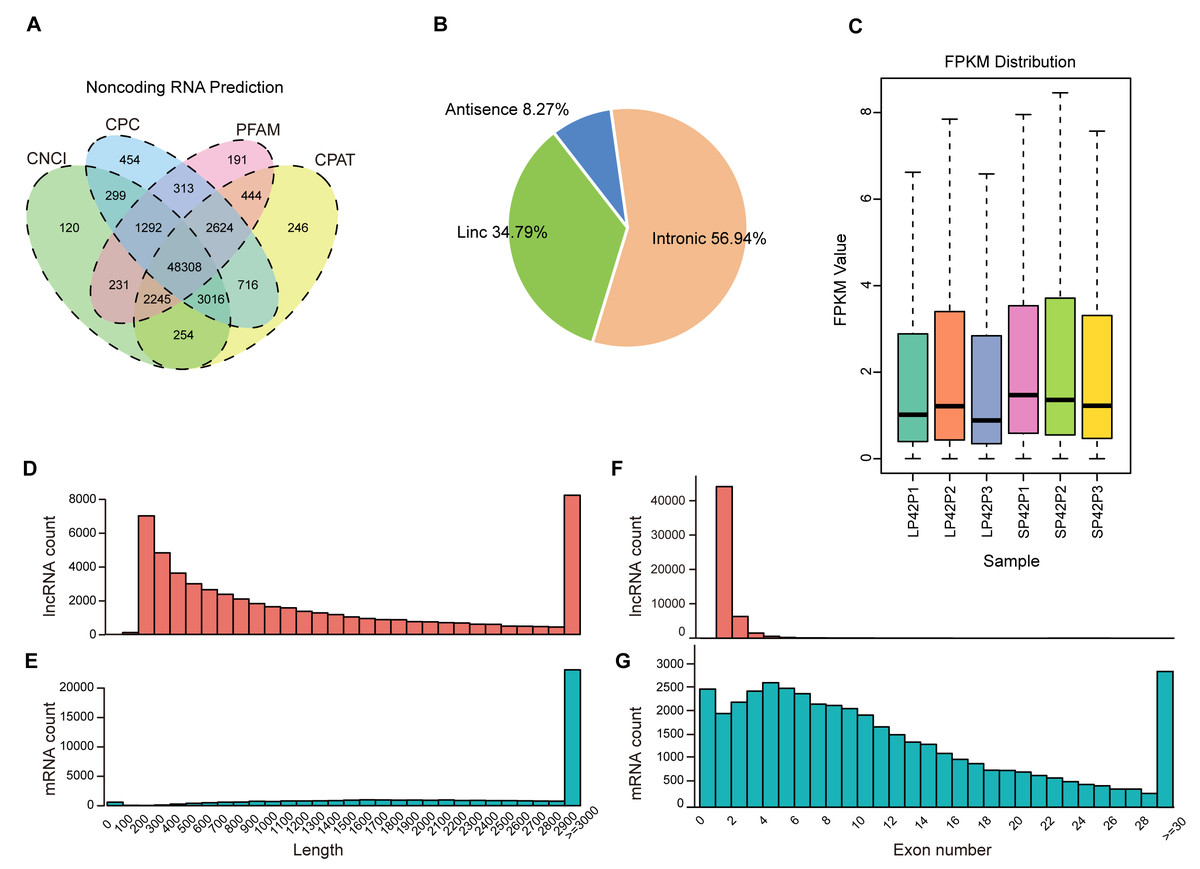
Figure 1: Identification of lncRNAs and mRNAs in ovine PD.
(A–B) The screen of lncRNAs. (C) Boxplot of FPKM distribution of each sample. (D–E) The length statistics of lncRNA and mRNA. (F–G) The statistics of lncRNA and mRNA exon number.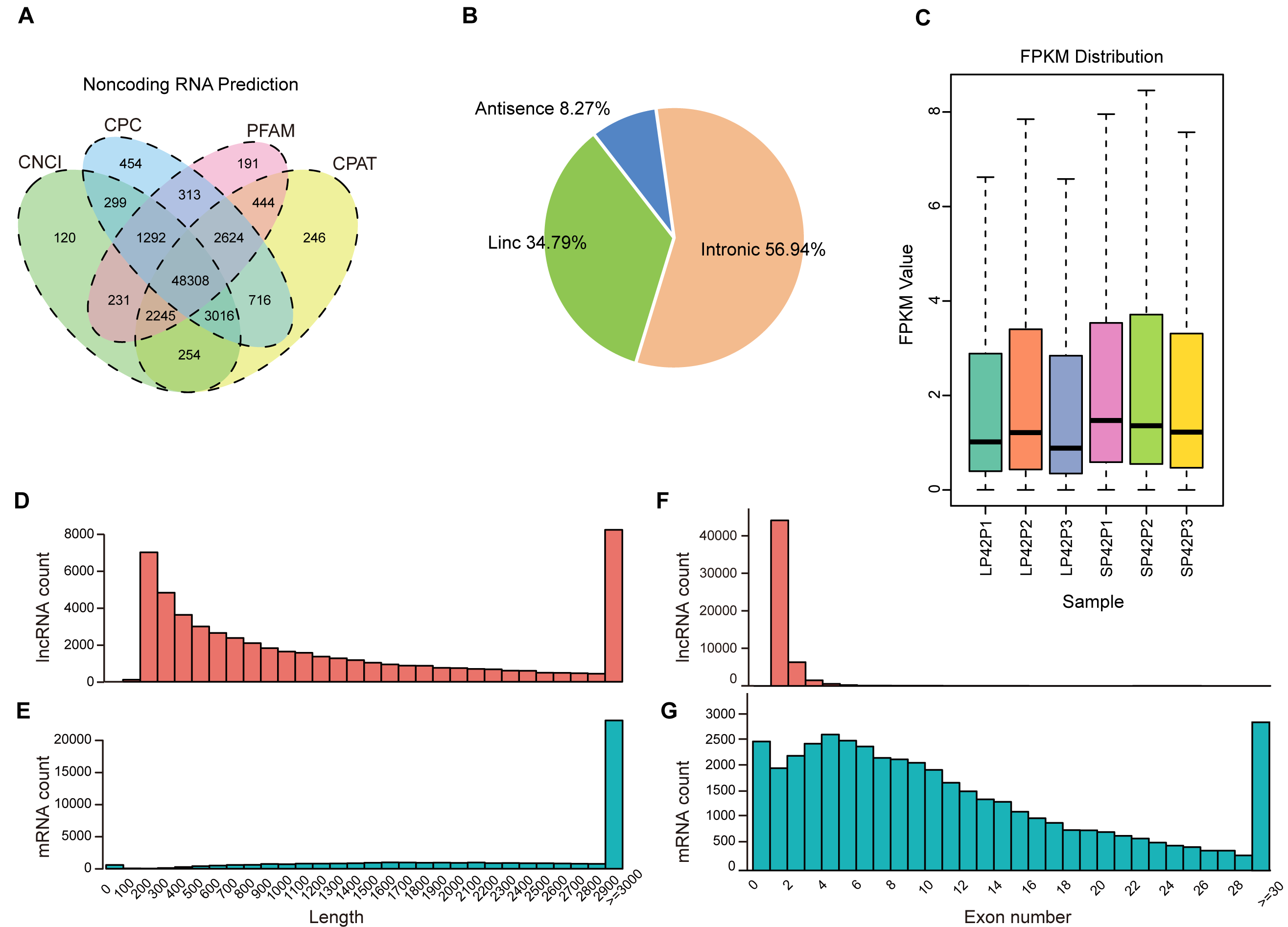{kind=link}
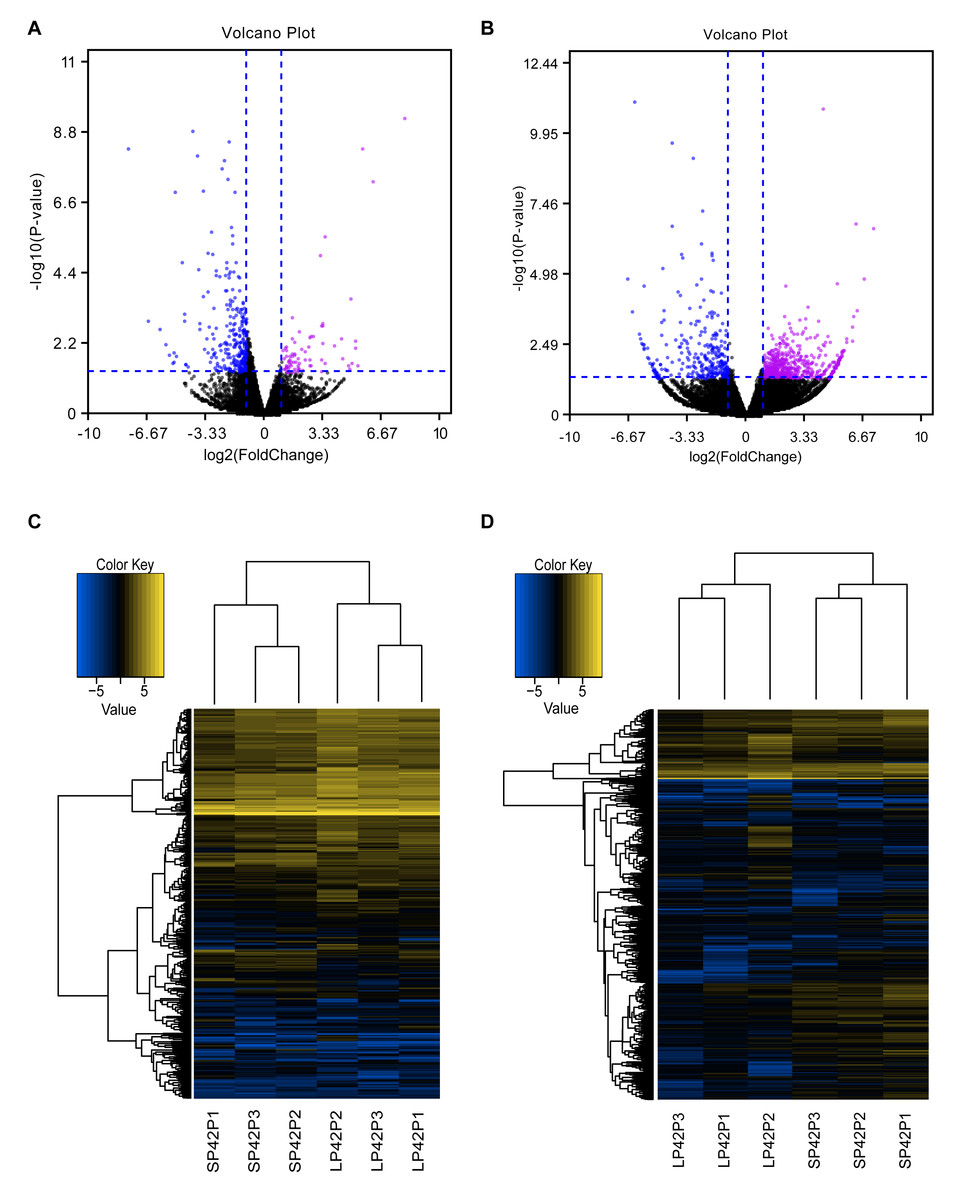
Figure 2: Analysis of differentially expressed transcripts.
(A) Volcano map of differentially expressed mRNA in SP42 and LP42. (B) Volcano map of differentially expressed lncRNA in SP42 and LP42. (C) Hierarchical cluster analysis of DE-mRNA in SP42 and LP42. (D) Hierarchical cluster analysis of DE-lncRNA in SP42 and LP42. The logarithm base 2 was taken to calculate Euclidean distance according to the expression of DE-mRNA and DE-lncRNA in each sample, then Hierarchical cluster maps were obtained.Note: purple and blue represent up-regulated and down-regulated transcripts respectively.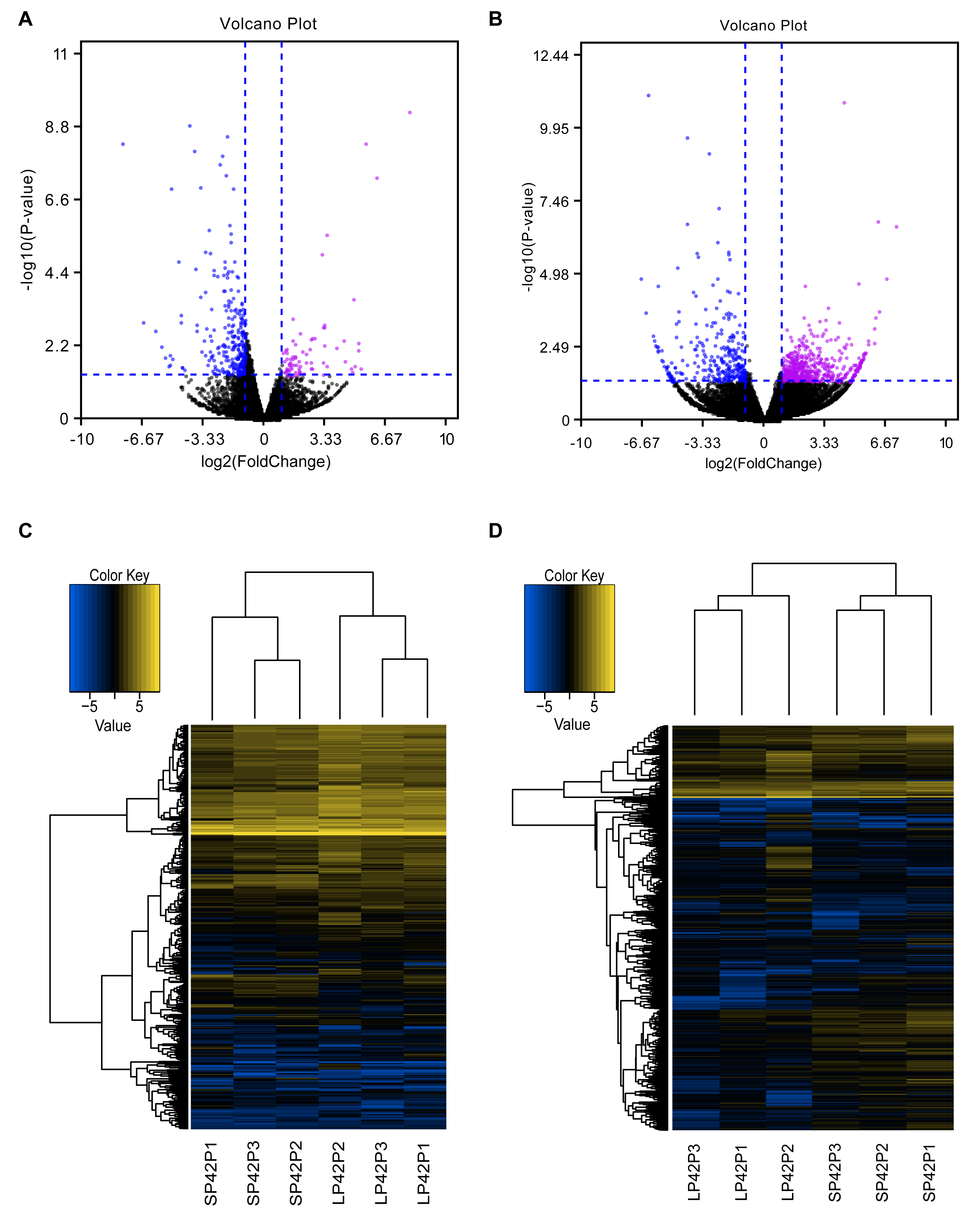{kind=link}
Analysis and verification of DE-lncRNAs and DE-mRNAs of ovine PD
In total, we identified 346 DE-lncRNA (Up-regulation 186 and down-regulation 163) and 186 DE-mRNA (Up-regulation 30 and down-regulation 156) comparing the SP42 to LP42 (Figs. 2A, 2B), the details of the up and down information can be found in Table S1 and Table S2. Besides, the hierarchical cluster analysis was performed to test the grouping is reasonable using DE-lncRNA and DE-mRNA (Figs. 2C, 2D). To verify the accuracy of sequencing, 5 DE-lncRNAs (MSTRG.251736, MSTRG.202202, MSTRG.26775, MSTRG.130282, MSTRG.50976) and 5 DE-mRNAs (DUSP6, ANGPTL2, SPRY4, GHRH, FOSL2) were selected randomly to detect the relative expression level in the SP42 and LP42 groups using qPCR. The expression levels of the 5 lncRNAs and 5 mRNAs were shown in Fig. 3 using the Lg(Relative expression), which were consistent with the RNA-seq both in lncRNA and mRNA.
Figure 3: Validation of the expression patterns of lncRNA and mRNA using qRT-PCR.
(A) The qPCR verification of the 5 DE-mRNAs in SP42 and LP42. (B) The qPCR verification of the 5 DE-lncRNAs in SP42 and LP42. The expression of transcripts was normalized by β-actin to determine relative expression using 2−ΔΔCt method. Results were expressed as mean±SE, * represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001.{kind=link}
GO and KEGG enrichment analysis of DE-mRNAs and DE-lncRNAs
We performed GO and KEGG enrichment analysis using the 346 DE-lncRNA and 186 DE-mRNA. The most significant (FDR < 0.05) enriched the top 10 terms of each GO type were shown in Fig. 4. For DE-mRNA, the significant enriched GO terms were involved in reproduction and PD function including response to the corticotropin-releasing hormone, negative regulation of ERK1, and ERK2 cascade, ion transport, regulation of type B pancreatic cell proliferation (Fig. 4A). For DE-lncRNA, we used their targets to conduct GO enrichment and most of the significant enriched GO terms participate in the regulation of biological and cellular processes (Fig. 4C). For KEGG enrichment analysis, the top20 enrichment pathways of DE-mRNA was shown in Fig. 4B, among them, pathways involved in reproductive hormone synthesis and secretion including oxytocin signaling, aldosterone synthesis, and secretion, insulin secretion, thyroid hormone synthesis, and GnRH signaling, as well as photoperiodic response pathway like Circadian entrainment. Moreover, the top20 enrichment pathways of DE-lncRNA targets showed that two reproduction associated pathways including Hippo and MAPK were significantly enriched (Fig. 4D).
Figure 4: GO and KEGG enrichment analysis of the DE-lncRNAs and DE-mRNAs in PD.
(A) GO function analysis of DE-mRNAs using the top 10 GO terms in each of BP, CC and MF. (B) Top 20 KEGG enrichment pathways of DE-mRNAs in ovine PD. (C) GO enrichment of DE-lncRNA targets using the top 10 GO terms in each of BP, CC and MF. (D) Top 20 KEGG enrichment pathways of DE-lncRNA targets in ovine PD. BP: Biological Process, CC: Cellular Component, MF: Molecular Function.{kind=link}
Screening of potential reproduction-related lncRNAs in ovine PD
In this part, firstly, 109 DE-lncRNA including 43 up-regulated and 66 down-regulated lncRNAs, as well as 93 DE-mRNA including 4 up-regulated and 89 down-regulated mRNAs were selected to construct the overall differentially expressed transcripts network (Fig. S1). The network showed that lncRNA was co-expressed with multiple protein-coding genes, which indicates mutual regulation of lncRNA and mRNA in the PD. Subsequently, a total of 20 DE-mRNAs, such as MAP2K3, TG, NR4A1, CREB3L1, CACNA1S, ADCYAP1R1, PER1, and a new gene LOC101104054, related to reproduction and photoperiodic response were used to construct the mRNA-mRNA network according to the KEGG enrichment results (Fig. 5A). To obtain the lncRNAs which participate in the ovine photoperiod induced reproductive activities, 40 lncRNAs, and its 11 targeted DE-mRNA which were discovered in the above mRNA-mRNA network were used to construct the interaction network. Surprisingly, the expression of these lncRNAs and mRNAs involved in sheep reproduction were all down-regulated in the PD (Fig. 5B). These findings suggest that PD tissue is greatly sensitive to in photoperiodic regulation of sheep reproduction.
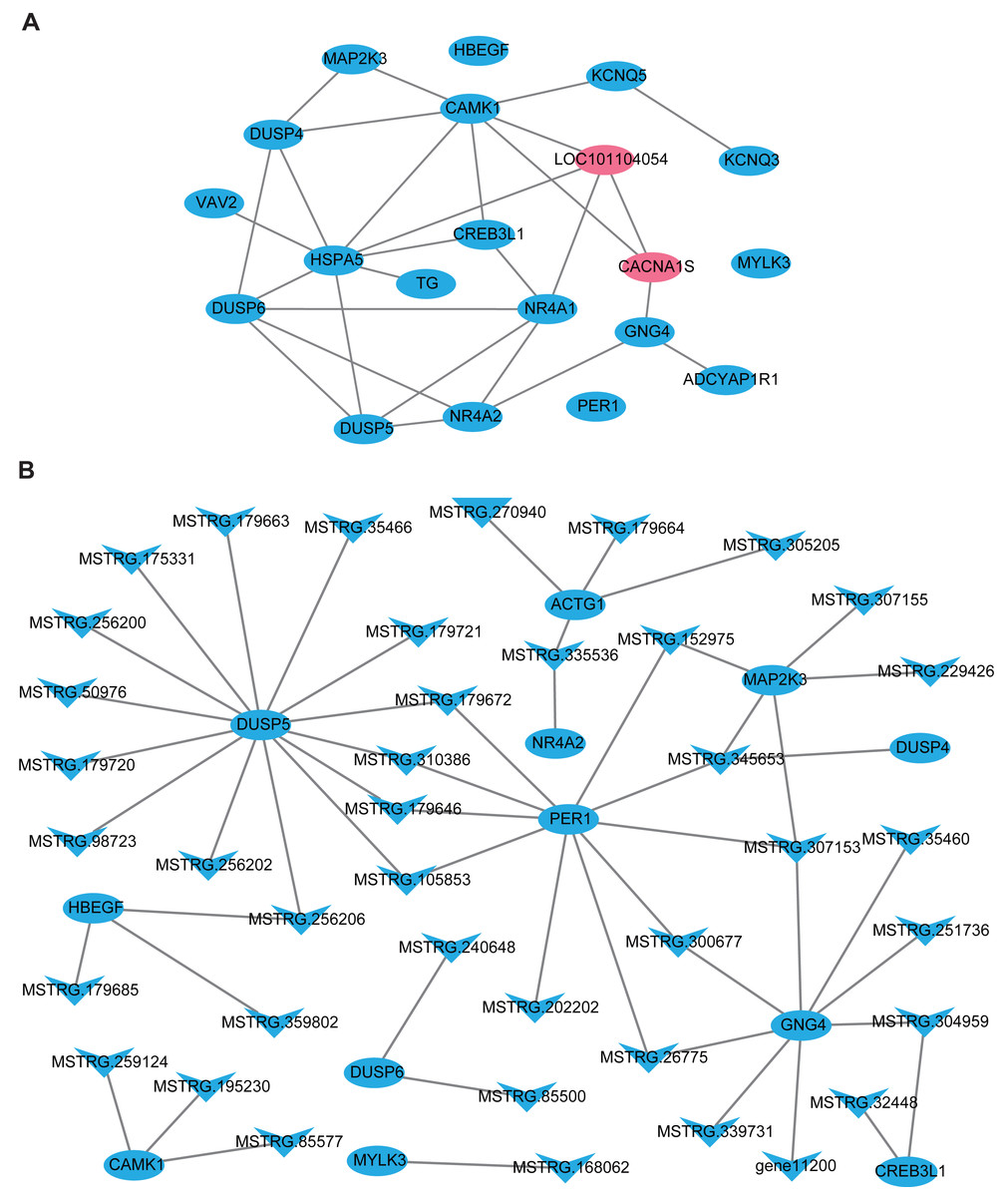
Figure 5: The network of differentially expressed transcripts involved in reproduction and photoperiodic response in the PD.
(A) The network of 20 DE-mRNAs involved in reproduction and photoperiodic response in the PD. (B) The network between DE-mRNAs with DE-lncRNAs involved in reproduction and photoperiodic response in the PD. Circles and “V” represent mRNAs and lncRNAs, purple and blue represent up-regulated and down-regulated transcripts respectively.{kind=link}
Discussion
The PD of pituitary is a crucial functional endocrine organ in the HPG axis that regulates mammalian onset of puberty and reproductive seasonality through the synthesis and secretion of reproductive hormones including FSH, LH and PRL. Recently studies have shown that synthesis and secretion of pituitary gonadotropins were regulated in the transcriptional level, in which noncoding RNA plays an important role (Yin et al., 2015; Lu et al., 2018; Pickard & Williams, 2016). Growing pieces of evidence indicate the important roles of lncRNAs in animal reproduction with the maturity of sequencing technology. For example, Liu found that several lncRNAs were involved in the reproductive related pathways such as TGF-β and PI3K-Akt by targeting the genes in the ovaries of Duroc pig (Liu et al., 2018), moreover, similar findings also exist in sheep (Feng et al., 2018; Wang et al., 2018), goat (Emanuele et al., 2018), chicken (Liu et al., 2018b) and other special economic animals (Chen et al., 2017; Sohel et al., 2013). In sheep, the lncRNAs in several tissues were related to animal reproductive or estrous activities (La et al., 2019; Zhang et al., 2019; Zheng et al., 2019; Feng et al., 2018). To better understand the effect of photoperiod on pituitary function, we established the OVX+E2 sheep model which has been frequently used to study the response to photoperiod and reproductive endocrine changes in seasonal reproduction animals (Legan et al., 2015; Scotti, Place & Demas, 2007; Jackson et al., 2013), subsequently, genome-wide analyses to identify differentially expressed mRNAs and lncRNAs in the pituitary of the above OVX+E2 sheep which treated with different photoperiod. The DE-lncRNAs and DE-mRNAs were used to reveal their functions in ovine pituitary, therefore this research provides a valuable resource for further studies of functional lncRNAs in the sheep pituitary.
Previous studies have proved that lncRNA was located in the protein-coding gene and can target this gene to play a regulatory role (Ulitsky et al., 2011; Guttman & Rinn, 2012). In this study, 48308 lncRNAs and 19906 mRNAs were identified in the pituitary of Sunite ewes, and the more lncRNAs were found compared with the earlier studies in the ovine pituitary (Zheng et al., 2019; Li et al., 2019b), suggesting that photoperiod may induce the production of more lncRNAs which participate in the regulation of pituitary function. The functional annotation of DE-lncRNA and DE-mRNA in case and control can reveal their roles clearly in a particular trait. In the present study, 20 of the DE-mRNAs (Fig. 5A), including MAP2K3, TG, NR4A1, CREB3L1, CACNA1S and ADCYAP1R1, were enriched in the pathways which were involved in the hormone synthesis and release, such as aldosterone synthesis and secretion, insulin secretion, thyroid hormone synthesis (Fig. 4B), and these pathways have been demonstrated to be involved in the mammalian reproductive regulation (La et al., 2019; Zheng et al., 2019; Li et al., 2019b). More interestingly, ADCYAP1R1 and PER1 were enriched in photoperiodic change-related pathway circadian entrainment. ADCYAP1R1 was the receptor gene of pituitary adenylate cyclase-activating polypeptide type 1 receptor (PAC1), Mercer et al. found that SNP rs2267735 within the ADCYAP1R1 located within a predicted estrogen response element, which regulates gene transcription when bound to estradiol (E2) activated estrogen receptor alpha (ER α) (Mercer et al., 2016). However, numerous pieces of evidence have confirmed that the concentration of E2was strongly affected by seasonal or photoperiodic change (Lai et al., 2012; Abdul-Rahman et al., 2016; Carr et al., 2003). PER1 is one of the typical clock genes, whose expression is significantly correlated with circadian rhythm (Albrecht & Eichele, 2003; Messager et al., 2001). Our findings indicated that photoperiod can change animal reproduction state through transcriptional regulation of key pathways in the mRNA level.
LncRNA can regulate the transcriptional activity of target genes and participate in organ function. In this study, we firstly constructed all the DElncRNA-DE mRNA interaction networks with the photoperiod change (Fig. S1), which shown that lncRNAs can regulate the expression of target genes through its up-down regulation. To more accurately search for the reproduction-related lncRNAs, we constructed the interactions of lncRNAs-mRNA according to the DE-mRNA related to reproduction which described above. Surprisingly, all of these transcripts were down-regulation. In general, SP stimulus induces secretion of thyrotropin (TSH) from the pars tuberalis (PT) of the anterior pituitary gland and this PT-derived TSH locally activates thyroid hormone (T3) within the hypothalamus, which in turn induces gonadotropin-releasing hormone (GnRH) and then gonadotropin secretion (Nakayama & Yoshimura, 2017). However, CREB3L1 can mediate functional and structural adaptation of the secretory pathway of thyroid hormone (García et al., 2017), MSTRG.32448 and MSTRG.304959 targeted CREB3L1 may be involved in the ovine reproduction by regulating the secretion of thyroid hormone. MAPK signaling has been reported to be associated with reproductive activity in sheep (Zheng et al., 2019), goat and rat (Gao et al., 2018), as well as there is accumulating evidence that extracellular signal-regulated kinase (ERK) is one of the mitogen-activated protein kinases (MAPKs), two ERK specific phosphatases DUSP5 and DUSP6, which have been demonstrated to be markers of MAPK signaling activation (Buffet et al., 2017). Moreover, the up-regulation of DUSP6 regulates the duration of ERK activation (Higa et al., 2018). This study suggested that lncRNA MSTRG.240648 and MSTRG.85500 may participate in the MAPK signaling activation through targeting DUSP6. Besides, 10 lncRNAs targeting PER1 gene, may relate to the photoperiod change, but the specific functions are still need systematic research.
Conclusion
In conclusion, transcriptome analyses are useful methods to understand the effects of photoperiodic change on seasonal reproduction in ewes. In this study, our differentially expressed analysis identified pivotal reproductive genes and lncRNAs in the pituitary, as well as their interaction relationship based on the OVX+E2 ewes. Function annotation analysis indicated that the reproductive hormone and photoperiod response-related pathways including oxytocin signaling, aldosterone synthesis and secretion, insulin secretion, thyroid hormone synthesis, and circadian entrainment were enriched in the pituitary. Several lncRNAs such as MSTRG.240648, MSTRG.85500, MSTRG.32448 and MSTRG.304959 targeted CREB3L1 and DUSP6 may be involved in the ovine reproductive activities. Besides, 10 lncRNAs may relate to the photoperiod change because of targeting PER1 gene. These findings in transcriptome provide a valuable resource for reproduction-related transcripts, and the interactions between DE-lncRNAs, DE-mRNAs and the enriched pathways provide clues for further study on the role of the pituitary regulated seasonal reproductive in ewes.
Supplemental Information
Supplemental figures and tables
Figure S1 The network between DE-mRNAs with DE-lncRNAs in the pituitary. Circles and “V” represent mRNAs and lncRNAs, line represent interaction between lncRNAs and mRNAs. Red and green represent up-regulated and down-regulated transcripts respectively.
Table S1Differentially expressed mRNA between the SP42 with LP42.
Table S2Differentially expressed lncRNA between the SP42 with LP42.
Table S3Top30 GO terms of differentially expressed mRNA between the SP42 with LP42.
Table S4Top30 GO terms of differentially expressed lncRNA targets between the SP42 with LP42.
Table S5Top20 differentially expressed mRNAs enriched KEGG pathways between the SP42 with LP42.
Table S6Top20 differentially expressed lncRNA targets enriched KEGG pathways between the SP42 with LP42.